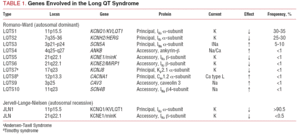
INTRODUCCIÓN
El síndrome de QT largo (SQTL) se caracteriza por una alteración grave de la repolarización ventricular, que da lugar a una prolongación del intervalo QT en el electrocardiograma (ECG). Esta enfermedad predispone a los pacientes a sufrir arritmias ventriculares malignas (torsade de pointes) y muerte súbita. La descripción clínica y electrocardiográfica del síndrome de QT largo fue comunicada en 1957 por Anton Jervell y Fred Lange Nielsen,1 que publicaron sus estudios sobre una familia de padres no consanguíneos con 6 hijos. Cuatro de los niños tenían sordera congénita y episodios sincopales, y 3 presentaron muerte súbita. El estudio del ECG de estos pacientes mostró un intervalo QT inusualmente largo. Ambos padres eran asintomáticos, tenían un ECG normal y no presentaban problemas de audición. En 1964, Romano y Ward comunicaron de forma independiente un síndrome cardíaco caracterizado por síncopes recurrentes, antecedentes familiares de muerte súbita y prolongación del intervalo QT sin sordera neuronal.2 Estudios genéticos posteriores demostraron que el síndrome descrito por Jervell y Lange Nielsen, que se acompaña de sordera neuronal congénita, corresponde a mutaciones homocigóticas, con un fenotipo grave y alto riesgo de muerte súbita. La afección conocida como síndrome de Romano-Ward corresponde generalmente a mutaciones heterocigotas, los pacientes no presentan alteraciones auditivas y la gravedad de la enfermedad varía considerablemente. Casi medio siglo después, en 1995,3,4 se describieron los principales genes asociados al SQTL y se reconoció la enfermedad como un trastorno de los canales iónicos cardíacos. Fue la primera canalopatía cardiaca que se describió y es quizá el trastorno arritmogénico de los canales iónicos que más se ha investigado hasta la fecha. El cuadro clínico es muy variable: el paciente puede ser asintomático o presentar síncopes recurrentes, convulsiones o muerte súbita como primera manifestación de la enfermedad. Inicialmente, el SQTL se consideraba un síndrome raro y, en efecto, la presentación grave de la enfermedad es esporádica. Sin embargo, la incidencia de las mutaciones relacionadas se estima en 1/3000-5000 casos,5 el 32% de los portadores asintomáticos pueden tener un intervalo QT corregido por la frecuencia cardíaca (QTc) dentro de los límites normales, la enfermedad se transmite al 50% de sus descendientes, son más susceptibles de desarrollar arritmias en comparación con la población general, y hasta el 20% pueden volverse sintomáticos.6
El síndrome de QT largo presenta una gran heterogeneidad genética. Se han descrito más de 500 mutaciones distribuidas en 10 genes en esta condición: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 y SCN4B. A pesar de los avances en este campo, no se puede establecer un diagnóstico genético en el 25%-30% de los pacientes.7,8 La presentación de la enfermedad es principalmente monogénica6; las variedades poligénicas o compuestas suelen tener un fenotipo más grave. La penetración, es decir, los pacientes que tienen la mutación y manifiestan el fenotipo, oscila entre el 25% y el 90%.9 Con menor frecuencia, puede haber variaciones en la expresividad de la enfermedad, con varios fenotipos resultantes de la misma mutación. Los estudios de genética molecular desarrollados en los últimos 11 años han aportado importantes correlaciones genotipo-fenotipo, que han ayudado a orientar el enfoque del tratamiento. Además, se han realizado interesantes observaciones sobre la susceptibilidad individual a desarrollar arritmias en los estudios que investigan los frecuentes polimorfismos no sinónimos en esta población, aspecto que ha despertado un considerable interés, especialmente en el área de la farmacogenómica.
CLASIFICACIÓN DEL SÍNDROME DEL QT LARGO
Conceptos generales
La clasificación del SQTL utilizada en el pasado se basaba en la presentación homocigótica o heterocigótica de la enfermedad, que da lugar al síndrome de Jervell-Lange-Nielsen (con sordera) y al síndrome de Romano-Ward (sin sordera), respectivamente. La presente clasificación hace hincapié en los hallazgos genéticos, como se ilustra en la Tabla 1. En 1995-1996 se describieron los tres principales genes asociados a la enfermedad. Estos genes, que codifican las unidades formadoras de poros de los canales de potasio IKs e IKr, y el canal de sodio Nav1.5, representan casi el 65% de los casos. Aunque en años posteriores se han incluido siete genes adicionales en la lista, sólo representan el 5% de los casos.
Los canales iónicos son proteínas transmembrana que transportan iones a través de la membrana celular. Los canales implicados en el SQTL son selectivos o especializados en el transporte de un solo ion y son dependientes de voltaje, es decir, su activación se produce a un voltaje intracelular específico, que varía según el subtipo de canal. Los fenómenos eléctricos y contráctiles que se producen en el cardiomiocito están controlados por estas estructuras. Los canales iónicos forman complejos macromoleculares compuestos por una unidad principal que forma el poro del canal y por proteínas auxiliares que lo regulan (Figura 1). La disfunción del canal observada en el SQTL puede producirse en estos dos sitios: la proteína principal o las proteínas reguladoras (Tabla 1). La afectación de la unidad formadora de poros, conocida como alfa, genera los tres subtipos más comunes de SQTL: SQT1 (que afecta al canal de potasio IKs), SQT2 (que afecta al canal de potasio IKr) y SQT3 (que afecta al canal de sodio). Al ser los subtipos más frecuentes, son los mejor caracterizados clínica y genéticamente. Las correlaciones fenotipo-genotipo en estas tres formas principales se describen en la figura 2. Actualmente, el síndrome de Jervell-Lange-Nielsen corresponde a las variedades 1 y 5 del SQTL. Característicamente, estos pacientes presentan sordera congénita y mutaciones compuestas homocigóticas o heterocigóticas que afectan a la corriente IKs. El síndrome de Romano-Ward incluye las variedades del SQTL 1 a 10 y no implica sordera.
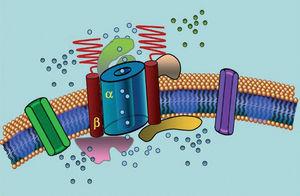
Figura 1. Representación esquemática del complejo macromolecular. Los canales iónicos son proteínas transmembrana (α) reguladas por varias proteínas; una de ellas es la llamada subunidad β.
Síndrome de QT largo tipo 1 (SQTL1)
Los pacientes con SQTL1 suelen presentar episodios de arritmia ventricular cuando hacen ejercicio o cuando se someten a un estímulo simpático (68%).10 Se ha descrito que la natación es un deporte desencadenante de arritmia en el SQTL1.11 La penetración es de casi el 62% en este subtipo. La onda T en estos pacientes suele tener una base ancha y una duración muy prolongada12,13 (Figura 2). Es el subtipo más frecuente y explica el 30%-35% de los casos. El gen afectado, KvLQT1 (o KCNQ1), está localizado en el cromosoma 11 (11p15.5) y codifica la subunidad α del canal de potasio IKs. El potencial de acción se prolonga por una reducción de la corriente de K+ saliente durante la fase 3 del potencial de acción.
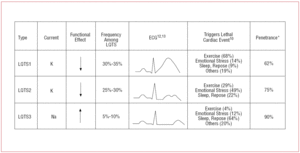
Figura 2. Correlación genotipo-fenotipo en los síndromes de QT largo más frecuentes. *Se refiere a los casos que tienen la mutación y manifiestan el fenotipo.
Síndrome de QT largo tipo 2 (SQTL2)
Los pacientes con SQTL2 tienden a presentar arritmia ventricular en respuesta al estrés emocional (49%) o a estímulos auditivos repentinos (p. ej., un reloj despertador), y con menor frecuencia durante el sueño (22%) o el ejercicio (29%).10 Las mujeres en el período posparto son especialmente susceptibles.14 La penetrancia estimada es del 79%; por lo tanto, hasta el 20% de los casos pueden tener un ECG no diagnóstico. La onda T en el SQTL2 suele ser de baja amplitud y bífida, con muescas12,13 (Figura 2). El gen afectado es el KCNH2 o HERG, localizado en el cromosoma 7 (7q35-36), que codifica la subunidad α del canal de potasio IKr y representa el 25%-30% de los casos. La disfunción de este canal disminuye la corriente de K+ saliente durante la fase 3 del potencial de acción, prolongando su duración.
Síndrome de QT largo tipo 3 (SQTL3)
Los pacientes con SQTL3 tienen un mayor riesgo de presentar arritmias malignas durante el reposo (sueño) o bradicardia.15 La penetrancia de la mutación del gen SCN5A es de casi el 90%. El ECG en el SQTL3 suele mostrar una onda T retrasada y puntiforme y permite observar claramente la prolongación del segmento ST12,13 (Figura 2). Estos pacientes suelen tener menos síntomas que los que padecen SQTL1 o SQTL2, pero los eventos son característicamente más letales.
El gen afectado en el SQTL3 es el SCN5A, que codifica la subunidad α del canal de sodio Nav1.5 (Figura 1), localizado en el cromosoma 3 (3p21-24); es la causa de la enfermedad en el 5%-10% de los casos. La inactivación defectuosa del canal permite la entrada sostenida de Na+ durante la fase 2 del potencial de acción, prolongando su duración.
El síndrome de QT largo tipo 4 (SQTL4)
El tipo 4 es una variedad rara del SQTL, que representa casi el 1% de los casos. Es una forma atípica que produce un amplio espectro de arritmias, incluyendo taquicardia ventricular polimórfica catecolaminérgica, fibrilación auricular, alteraciones de la conducción intraventricular, disfunción del nodo sinusal y bradicardia6-18; además, el QTc puede estar dentro de los límites normales en muchos pacientes. El gen afectado es el ANKB, localizado en el cromosoma 4 (4q25-27), que codifica la síntesis de la anquirina-β, una proteína estructural que une las proteínas de la membrana de los cardiomiocitos a las proteínas del citoesqueleto. Estas proteínas son la bomba Na/K ATPasa, el intercambiador Na/Ca y el receptor de inositol trifosfato (InsP3R). Las mutaciones que provocan una pérdida de la función de la ankirina-β conducen a un aumento de la concentración de calcio intracelular y a alteraciones en la expresión de la N/K ATPasa y del intercambiador Na/Ca. La elevada concentración de calcio da lugar a posdespolarizaciones tempranas y retardadas. Así pues, las arritmias ventriculares observadas en las mutaciones del gen de la anquirina-β se deben a despolarizaciones espontáneas, generalmente en respuesta a la estimulación catecolaminérgica.
El síndrome de QT largo tipo 5 (SQTL5)
El tipo 5 se origina con cambios en la secuencia del gen KCNE1 localizado en el cromosoma 21 (21q22.1p22.)19 KCNE1 codifica la síntesis de la subunidad β del canal IKs, también conocida como subunidad minK, que regula el canal IKs. Este tipo representa menos del 1% de los casos.
Síndrome de QT largo tipo 6 (SQTL6)
El gen afectado en el tipo 6 es el KCNE2, situado en el cromosoma 21 (21q22.1).20 Este gen codifica la subunidad β del canal de potasio, también conocida como subunidad MiRP1, y regula el canal IKr. Menos del 1% de los casos son del tipo 6.
El síndrome de QT largo tipo 7 o síndrome de Andersen-Tawil (SQTL7)
Los hallazgos dismórficos y las alteraciones electrocardiográficas que se observan en este síndrome fueron descritos por primera vez en 1971 por el Dr. Andersen21 y revisados en 1994 por el Dr. Tawil,22 pero la descripción genética/molecular no se comunicó hasta 2001.23 Ahora conocida como síndrome de Andersen-Tawil (ATS), esta condición es una alteración autosómica dominante caracterizada por parálisis periódica, desarrollo esquelético anormal, arritmia ventricular del tipo de extrasístoles ventriculares frecuentes, y una particular susceptibilidad a desarrollar fibrilación ventricular, particularmente en mujeres. Las alteraciones descritas en la ETA incluyen extrasístoles ventriculares (41%), taquicardia ventricular polimórfica no sostenida (23%), taquicardia ventricular bidireccional (68%) y torsade de pointes (3%).24 Algunas de las características dismórficas observadas incluyen baja estatura, escoliosis, clinodactilia, hipertelorismo, implantación baja de las orejas, micrognatia y frente ancha. La expresión de la enfermedad es variable, hecho que complica el diagnóstico precoz.23,25 Las mutaciones en el gen KCNJ2 situado en el cromosoma 17 (17q23), que codifica la síntesis del canal de potasio rectificador Kir 2.1, son responsables del 70% de los casos. Este canal participa en la fase 4 del potencial de acción. Varios autores cuestionan la inclusión de este gen dentro del grupo causal del SQTL, porque el intervalo QTc sólo está ligeramente prolongado en este síndrome o incluso es normal, pero la onda U suele ser prominente, lo que ha llevado a sobreestimar el intervalo QT. El lector encontrará que algunos autores sugieren que las mutaciones de KCNJ2 generan el ATS1 y no el LQTS7.24
El síndrome de QT largo tipo 8 (LQTS8)
El tipo 8 surge de mutaciones en el gen CACNA1 situado en el cromosoma 12 (12p13.3), que codifica el canal de calcio tipo L Cav1.2. Provoca el síndrome de Timothy,26 una afección caracterizada por malformaciones cardíacas, deficiencia inmunológica intermitente, hipoglucemia, alteraciones cognitivas, incluido el autismo, fusión interdigital y prolongación del QT, que provoca arritmia cardíaca y muerte súbita.27 Menos del 0,5% de los casos son del tipo 8.
Síndrome de QT largo tipo 9 (SQTL9)
Esta variedad de SQTL se desarrolla a partir de mutaciones en el gen CAV3, situado en el cromosoma 3 (3p25), que codifica la síntesis de la caveolina 3. La caveola es una invaginación de la membrana plasmática implicada en la endocitosis, la homeostasis lipídica y la transducción de señales. Un componente importante de esta estructura es la caveolina, que tiene 3 subtipos conocidos; el subtipo 3 es específico del músculo esquelético y cardíaco. Algunos canales iónicos están ubicados en la caveola, incluida una isoforma cardíaca del canal de sodio Nav1.5. Recientemente se han descrito varias mutaciones en esta proteína. Éstas alteran las propiedades biofísicas del canal de sodio Nav1.5 in vitro, generando un fenotipo similar al observado en el SQTL3.28 Menos del 1% de los casos se atribuyen a esta causa.
El síndrome de QT largo tipo 10 (SQTL10)
El tipo 10 se describió en un caso muy grave, con QTc >600 ms, bradicardia fetal y bloqueo auriculoventricular (AV) 2:1. Es el resultado de mutaciones en el gen SCN4B, situado en el cromosoma 11 (11q23), que codifica la subunidad β4 del canal de sodio. Se han descrito cuatro subtipos diferentes de subunidades β, que interactúan y regulan las distintas isoformas del canal de sodio; sin embargo, hasta ahora sólo el subtipo 4 se ha asociado a la arritmogénesis.29 La incidencia de las mutaciones de este gen no ha sido examinada, pero se estima en
Mutaciones de la variedad Jervell-Lange-Nielsen
Esta forma grave de SQTL está causada por mutaciones homocigóticas30 o heterocigóticas compuestas de los genes KCNQ1, y/o KCNE1, que codifican la corriente IKs; es decir, una variedad muy grave de las formas LQTS1 o LQTS5. Esta enfermedad se asocia característicamente a la sordera congénita. Los pacientes suelen tener un QTc>500 ms y síncopes recurrentes, y tienen un alto riesgo de muerte súbita. Los padres de los pacientes con esta variedad suelen ser heterocigotos y tienen una enfermedad menos grave, o no muestran síntomas.31
DIAGNÓSTICO DEL SÍNDROME DEL QT LARGO
Puntuación de Schwartz
En 1985, Schwartz et al32 publicaron los criterios para diagnosticar el SQTL, que fueron modificados en 1993 y contienen importantes pautas para la evaluación inicial de los posibles casos. Este sistema utiliza una puntuación de 1 a 9 basada en los antecedentes familiares y los hallazgos clínicos y electrocardiográficos. La probabilidad de enfermedad es baja con una puntuación de ≥1, intermedia con 2-3 y alta con ≥4 (tabla 2).

Diagnóstico prenatal del síndrome de QT largo
La bradicardia fetal puede ser una de las primeras manifestaciones clínicas del SQTL. Series retrospectivas han demostrado que hasta el 70% de los pacientes diagnosticados de SQTL durante la infancia tienen una historia de bradicardia, normalmente acompañada de hidropesía fetal.33 La evaluación de la repolarización cardíaca fetal entre las semanas 14 y 39 es útil para el diagnóstico precoz del SQTL.34
El mosaicismo gonadal para el SQTL se ha asociado con pérdidas fetales recurrentes durante el tercer trimestre del embarazo.35 Si se sospecha mucho de la enfermedad, la amniocentesis después de las 16 semanas de gestación puede ser útil para establecer el diagnóstico, al que se llega fácilmente cuando se sabe que uno de los padres es portador de una mutación específica.36
Estudio de un paciente con síndrome de QT largo
Historia clínica
Los antecedentes familiares y/o personales de muerte súbita son de crucial importancia tanto para el diagnóstico como para la estratificación del riesgo del SQTL. Además, los factores precipitantes y el contexto del síncope pueden indicar el subtipo de SQTL. En la evaluación inicial de un caso sospechoso, debe descartarse el uso de fármacos que puedan prolongar el intervalo QT.
Intervalo QT: ¿Qué es normal?
El intervalo QT debe medirse preferentemente en las derivaciones II o V5,37 donde se ha demostrado que tiene mayor valor predictivo.38 Este intervalo indica la duración de la repolarización ventricular y se mide desde el inicio de la onda Q hasta el final de la onda T. Convencionalmente se emplea la fórmula propuesta por Bazett39 para corregir la duración del intervalo en función de la frecuencia cardíaca (QTc=QT/√RR, expresado en segundos). Aunque la medición del intervalo QT parece sencilla, en un estudio multicéntrico realizado por Viskin et al,40 menos del 40% de los médicos que no son cardiólogos, menos del 50% de los cardiólogos y más del 80% de los especialistas en arritmias sabían medirlo correctamente. Es aconsejable que los médicos realicen la medición manual y no confíen en las mediciones automatizadas, que pueden ser útiles para otros intervalos, pero son imprecisas al calcular el intervalo QT. El QT es un intervalo dinámico y los límites normales dependen de varios factores. Aunque un intervalo QTc de é440 ms en varones y é460 ms en mujeres se considera anormal, se pueden encontrar tanto portadores de mutaciones como individuos sanos dentro de este rango (Figura 3). En familias con SQTL1, Vincent et al41 demostraron que ninguno de los casos con genotipo positivo tenía un QTc470 ms. Monnig et al38 han demostrado recientemente que el QTc>440 ms es suficiente para detectar a los pacientes con mutaciones asociadas al SQTL, el QTc>470 ms es útil para identificar a los pacientes con riesgo de desarrollar síntomas, y el QTc>500 ms se encuentra en los pacientes sintomáticos sometidos a tratamiento.
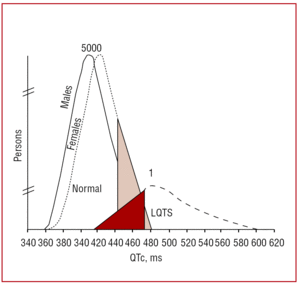
Figura 3. Modelo que muestra la distribución del intervalo QT corregido por la frecuencia cardíaca (QTc) en pacientes con mutaciones en KVLQT1, HERG o SCN5A, y sus familiares no afectados. La curva de la izquierda describe la distribución de los miembros no afectados y la curva de la derecha, la de los miembros afectados.
Otras alteraciones electrocardiográficas asociadas al síndrome de QT largo
Los pacientes con SQTL pueden presentar múltiples alteraciones de la onda T: alternancia de polaridad, variaciones de amplitud, muescas y aspecto bifásico, entre otras.42 La alternancia de la onda T (figura 4A) se define como una variación latido a latido de la amplitud, la morfología y la polaridad de una onda T de ritmo sinusal, sin variaciones en el complejo QRS. Es un indicador de inestabilidad eléctrica,43 que refleja la dispersión regional de la repolarización ventricular, y ocasionalmente precede a la fibrilación ventricular.44
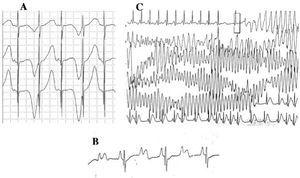
Figura 4. Alteraciones electrocardiográficas en el síndrome de QT largo. A: alternancia eléctrica de la onda T. B: bloqueo auriculoventricular 2:1. C: torsade de pointes autolimitada.
Los pacientes con SQTL pueden evolucionar con signos de disfunción del nodo sinusal, bradicardia y/o pausas.45 Los subtipos de SQTL1 y SQTL3, sobre todo este último, suelen presentar bradicardia sinusal,46 mientras que el SQTL4 se ha asociado a la disfunción del nodo sinusal.18
Desde la década de 1970-1980, se ha observado la coexistencia de defectos de conducción AV con el SQTL47 (figura 4B). El bloqueo AV dos a uno es una manifestación poco frecuente y de mal pronóstico que puede estar presente desde la etapa fetal en forma de bradicardia persistente. La incidencia de esta anomalía se ha descrito en un 4%-5%48 y se asocia a una elevada mortalidad a pesar del tratamiento con betabloqueantes y/o marcapasos.49,50 Este fenómeno puede explicarse por una duración prolongada del potencial de acción. Cuando el período refractario ventricular se prolonga, el siguiente impulso de actividad sinusal se bloquea porque llega a los ventrículos cuando todavía están en el período refractario. Esta alteración parece ocurrir exclusivamente en el SQTL, porque el período refractario ventricular es mayor que el del sistema de conducción AV.51 La pendiente del complejo QRS suele ser pronunciada y el bloqueo se ha localizado en la zona infrahisiana,46,51,52 pero el lugar del bloqueo puede depender del genotipo. Hasta ahora, se han relacionado 4 genes con el bloqueo 2:1 en el SQTL: HERG (SQTL2),53,54 SCN5A (SQTL3),52 CACNA1 (SQTL8),26 y SCN4B (SQTL10).55
La arritmia ventricular característica del SQTL se conoce como torsade de pointes (Figura 4C). Se presenta cuando el intervalo QT está prolongado, independientemente de la etiología. Es una taquicardia ventricular polimórfica debida a reentrada, caracterizada electrocardiográficamente por un giro continuo del eje del QRS alrededor de una línea imaginaria. Suele ir precedida de una pausa seguida de una extrasístole (intervalo RR corto-largo-corto), como se muestra en la figura.56-58 Puede culminar en fibrilación ventricular y muerte súbita. Si esto no ocurre, el paciente puede experimentar sólo un síncope, y si el episodio es breve, puede pasar desapercibido.
El estudio Holter
proporciona una evaluación completa y dinámica del intervalo QT. Ocasionalmente se registran episodios espontáneos de arritmia ventricular asintomática, así como episodios de disfunción del nódulo sinusal o de bloqueo AV.
Prueba de esfuerzo
Los pacientes con SQTL no pueden alcanzar la frecuencia cardíaca máxima esperada calculada según la edad. Además, bajo esfuerzo el intervalo QT puede mostrar un comportamiento paradójico, al aumentar en lugar de disminuir.59,60 El patrón electrocardiográfico durante la prueba de esfuerzo será diferente dependiendo del tipo de SQTL. Los pacientes con SQTL1, además de no alcanzar la frecuencia cardíaca máxima calculada para su edad, muestran con frecuencia un aumento del intervalo QT, mientras que aquellos con SQTL2 pueden alcanzar su frecuencia cardíaca esperada y mostrar sólo un leve aumento del intervalo QT o ninguno.61,62 En general, los pacientes con SQTL3 tienen una respuesta fisiológica al ejercicio, es decir, un acortamiento normal del intervalo QT.63 Las pruebas de esfuerzo también pueden ser útiles para evaluar la respuesta al tratamiento y para estratificar el riesgo en los casos asintomáticos, o cuando existen dudas sobre los eventos que conducen a la arritmia.
Cribado genético
En los últimos años, los estudios genéticos en el SQTL se han limitado a los laboratorios de investigación. Sin embargo, la información derivada de estos esfuerzos ha sido de gran utilidad para el tratamiento de los pacientes, especialmente de los casos de alto riesgo. Quizá la principal aplicación del cribado sea el asesoramiento genético, pero también tiene importantes implicaciones en el tratamiento, que puede orientarse en función del canal afectado. La localización precisa de una determinada mutación puede aportar información adicional sobre la evolución del riesgo. Los pacientes con mutaciones en la región transmembrana de KCNQ1 (IKS) tienen una mayor probabilidad de presentar eventos arrítmicos que aquellos con mutaciones en la región C-terminal64; lo mismo ocurre con los pacientes con mutaciones en la región del poro de KCNH2 o HERG65 en comparación con aquellos con mutaciones en las regiones N- o C-terminal66.
El cribado inicial puede limitarse quizás a los genes KCNQ1, HERG y SCN5A, que ofrecen la posibilidad de encontrar mutaciones en el 65% de los casos. Cuando los resultados obtenidos son negativos, el cribado puede ampliarse a los genes KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 y SCN4B, lo que aumentará la posibilidad de obtener resultados positivos entre un 5% y un 10%.
Cribado genético postmortem
Es interesante que se hayan encontrado mutaciones genéticas que conducen al SQTL en niños que experimentaron una muerte súbita y en casos inexplicables de muerte súbita en adultos jóvenes.
Los estudios genéticos postmortem de pacientes con muerte súbita y autopsia negativa han mostrado mutaciones que conducen al SQTL en porcentajes variables67-69: cerca del 10% en niños y el 35% en adultos jóvenes.70-72 En base a estos resultados, se ha propuesto el estudio rutinario del ECG en todos los recién nacidos.73,74
El estudio genético postmortem, también conocido en la literatura como «autopsia molecular», además de las repercusiones legales, tiene importantes implicaciones en las familias que pueden estar afectadas sin que lo sepan.
Polimorfismos reguladores
Se han descrito varios polimorfismos de aparición frecuente en la población con SQTL, distribuidos en casi todos los genes asociados a esta condición. Aunque estos cambios no son aparentemente patógenos, algunos pueden tener los siguientes efectos75-78:
1. Generar susceptibilidad individual a desarrollar arritmias.
2. Favorecer el impacto patogénico de otro cambio no sinónimo.
3. Disminuir el efecto patogénico de otro cambio no sinónimo.
Este es el caso del polimorfismo K897T en KCNH2 (HERG), que está presente hasta en el 15% de la población y que no sólo se relaciona con la susceptibilidad a ciertos fármacos,79 sino que también favorece el efecto patogénico de mutaciones en el mismo gen.78 Otro ejemplo es el polimorfismo S1103Y en el gen SCN5A, que se encuentra principalmente en personas de raza negra, que tiene una incidencia de casi el 13% y se asocia con un mayor riesgo de muerte súbita en la infancia.80
Curiosamente, se han descrito dos sitios de procesamiento alternativo que generan dos tipos de canales de sodio en el producto del gen SCN5A, (que codifica la isoforma del canal de sodio Nav1.5 en los humanos): uno con 2016 aminoácidos que contienen glutamina en la posición 1077 (Q1077), y otro con 2015 aminoácidos que carecen de glutamina (Q1077del). Los transcritos de estas transformaciones alternativas están presentes en una proporción 2:1 en el mismo corazón humano y varios polimorfismos frecuentes tendrán efectos diferentes en el funcionamiento del canal, dependiendo de si el contexto es Q1077 o Q1077del. Esto se demostró inicialmente con el polimorfismo H558R del SCN5A, presente hasta en el 30% de la población. Cuando el H558R se expresó en el contexto de Q1077, se observó una profunda reducción de la corriente iónica.81 Un efecto similar se documentó con el polimorfismo S524Y82. Estos hallazgos han proporcionado factores para explicar la gravedad variable de la enfermedad, así como los diferentes fenotipos de la misma mutación observados en algunas familias.77
Pruebas farmacológicas con adrenalina
Las pruebas farmacológicas con dosis bajas de adrenalina son una opción útil y segura para desenmascarar los casos sospechosos de SQTL con un QTc límite. Es especialmente eficaz para detectar formas asintomáticas de SQTL1, con una sensibilidad del 92,5%, una especificidad del 86%, un valor predictivo positivo del 76% y un valor predictivo negativo del 96%. También puede ser útil en el diagnóstico del SQTL2, con una sensibilidad y especificidad menores. No es útil para el SQTL3 u otras formas de SQTL. En condiciones normales, la estimulación simpática induce la fosforilación del canal de potasio IKs, optimizando su función y dando lugar a un acortamiento del potencial de acción. En pacientes con SQTL, en particular el tipo 1, se observa una respuesta paradójica a la administración de dosis bajas de adrenalina (0,025-0,2 µg/kg/min) que prolonga el intervalo QT a más de 30 ms83-86.
PROLONGACIÓN DEL INTERVALO QT Y TORSADE DE PUNTOS INDUCIDA POR FÁRMACOS
Una gran variedad de fármacos utilizados en diferentes especialidades médicas pueden provocar un aumento iatrogénico del intervalo QT. Algunos fármacos han sido retirados del mercado debido a este efecto indeseable (por ejemplo, astemizol y cisaprida, entre otros; para más información, visite www.qtdrugs.org).87,88
La arritmia ventricular secundaria a fármacos no antiarrítmicos se produce en menos de uno de cada 10 000 a 100 000 sujetos expuestos. Teniendo en cuenta que los estudios clínicos incluyen entre 2000 y 3000 sujetos, este indeseable y fatal acontecimiento adverso escaparía fácilmente a la detección durante la fase clínica de desarrollo del fármaco.89 Este punto ha generado un enorme interés en los aspectos referidos a la seguridad en el estudio y desarrollo de nuevos fármacos.
Los factores relacionados con la susceptibilidad individual incluyen el sexo femenino, la hipocalcemia, la hipomagnesemia, la bradicardia, la insuficiencia cardíaca, la postcardioversión, la fibrilación auricular, la hipertrofia ventricular izquierda, el SQTL no detectado, los polimorfismos predisponentes y las concentraciones séricas elevadas de fármacos predisponentes.90
El canal que suele interactuar con los fármacos es el IKr, codificado por el gen KCNH2(HERG), debido a su estructura molecular. Otros canales de potasio tienen 2 residuos de prolina angulados hacia el poro del canal, reduciendo su lumen. En cambio, el IKr carece de estos residuos, se genera un vestíbulo de poro más grande y se facilita la exposición a moléculas grandes. Además, tiene 2 residuos aromáticos (tirosina y fenilalanina) que favorecen la unión con moléculas aromáticas presentes en varios fármacos capaces de bloquear el canal.91
Como se mencionó anteriormente, la penetrancia del SQTL es incompleta y algunos portadores asintomáticos de mutaciones podrían manifestar arritmia maligna al recibir uno de estos fármacos. Además, los polimorfismos considerados frecuentes en la población confieren una susceptibilidad individual al desarrollo de torsade de pointes cuando se utilizan algunos fármacos. Este es el caso del polimorfismo R1047L, el segundo más frecuente en KCNH2, que se ha asociado al desarrollo de torsade de pointes con el uso del fármaco dofetilida.92 Se han descrito al menos 20 polimorfismos del gen KCNH2 en personas sanas y su efecto en la susceptibilidad individual a desarrollar arritmias relacionadas con fármacos está por determinar.93 También se han documentado polimorfismos que confieren susceptibilidad al desarrollo de arritmias ventriculares en el canal de sodio Na1.5. Este es el caso del polimorfismo H558R, que está presente hasta en el 30% de la población, o el S1103Y, que es frecuente en la población negra80,81,90,94,95; su implicación en la susceptibilidad inducida por fármacos no ha sido investigada.
SÍNDROME DEL QT LARGO Y EMBARAZO
El consejo genético es importante en el SQTL, pero en términos generales no hay contraindicación para el embarazo en mujeres portadoras, aunque cada caso es diferente y debe ser valorado individualmente en el contexto adecuado.
Se ha observado que el riesgo de presentar arritmia ventricular maligna disminuye con el embarazo. Por el contrario, se ha descrito una mayor vulnerabilidad a presentar arritmia maligna en los primeros 9 meses tras el parto, especialmente en pacientes con SQTL2. Este riesgo disminuye considerablemente con el tratamiento con betabloqueantes.96
ESTRATIFICACIÓN DEL RIESGO
La evolución del SQTL es variable y está influida por la duración del intervalo QTc, los factores ambientales, la edad, el genotipo y la respuesta al tratamiento.97,98 La arritmia ventricular es más frecuente en el SQTL1 y en el SQTL2, pero es más grave en el SQTL3.99 Como se ha mencionado anteriormente, las mujeres son especialmente susceptibles de sufrir arritmias malignas durante el período posparto.14
El síndrome de QT largo debe considerarse de alto riesgo cuando se asocia a lo siguiente:
1. Sordera congénita 1. Sordera congénita (síndrome de Jervell-Lange-Nielsen). Síncope recurrente debido a una taquiarritmia ventricular maligna.
3. Antecedentes familiares de muerte súbita.
4. QTc>500 ms.
5. Bloqueo auriculoventricular 2:1.
6. Alternancia eléctrica de la onda T.
7. Genotipo del SQTL3.
El estudio de Priori et al97 realizado en 647 pacientes demostró que la probabilidad de presentar un evento mayor (síncope, paro cardíaco, muerte súbita) antes de los 40 años es alta (>50%) cuando el QTc es >500 ms en el SQTL1, el SQTL2 y en los varones con SQTL3. Recientemente, se ha publicado un análisis del registro internacional de SQTL. Se analizó el riesgo de muerte súbita en 2772 adolescentes con la enfermedad, y se identificaron 3 factores asociados a un mayor riesgo en esta población: QTc>530 ms, historia de síncope en los últimos 10 años y sexo; los niños de 10 a 12 años tenían un riesgo mayor que las niñas, pero en el rango de edad de 13 a 20 años, el riesgo era comparable.100
TRATAMIENTO
Los pacientes sintomáticos que no reciben tratamiento tienen una tasa de mortalidad anual del 20% y una mortalidad a 10 años del 50% tras un primer evento de arritmia ventricular. Aunque está claro que el tratamiento debe establecerse cuando hay síntomas, el enfoque a utilizar en pacientes asintomáticos sigue siendo objeto de debate. Se ha documentado que la parada cardiaca puede ser la primera manifestación de la enfermedad en el 9% de los pacientes48 , y que el 12% de los pacientes asintomáticos desarrollarán síntomas y pueden experimentar una muerte súbita. El tratamiento inicial con betabloqueantes debe iniciarse en todos los pacientes con SQTL. La restricción del ejercicio es recomendable, pero los marcadores de riesgo clínicos y electrocardiográficos son una base útil para la toma de decisiones. Es importante informar a los pacientes sobre el riesgo de utilizar varios fármacos que pueden prolongar el intervalo QT y favorecer el desarrollo de arritmias ventriculares, como se ha mencionado anteriormente. El diagnóstico genético, además de permitir un adecuado asesoramiento familiar relacionado con la enfermedad, es una ayuda para valorar el pronóstico y orientar el tratamiento específico.
Los betabloqueantes
son el tratamiento de primera línea para el SQTL y todos los pacientes deben recibirlos como terapia inicial.101 Proporcionan una reducción del riesgo de eventos cardiovasculares de hasta el 64%100 y son especialmente eficaces en pacientes con mutaciones de los canales IKs (SQTL1),102 que están regulados en gran medida por el sistema simpático. Los betabloqueantes no modifican el intervalo QT, sino su dispersión.103 Aunque estos fármacos disminuyen la incidencia de eventos,104,105 se ha demostrado que el 10% de los pacientes con SQTL1, el 23% con SQTL2 y el 32% con SQTL3 tendrán síntomas cardiovasculares a pesar del tratamiento.106 Los pacientes con SQTL3, en particular, no parecen obtener beneficios importantes; de hecho, este grupo de fármacos debe utilizarse con precaución en estos pacientes, porque los episodios de arritmia ventricular en el SQTL3 son más frecuentes cuando la frecuencia cardíaca es baja. En términos generales, el 32% de los pacientes sintomáticos tendrán síntomas recurrentes en los primeros 5 años antes de iniciar el tratamiento con betabloqueantes, y el 14% de los pacientes rescatados de un episodio de muerte súbita presentarán otro evento similar en los siguientes 5 años si sólo reciben esta terapia.107 Se han utilizado varios betabloqueantes en el tratamiento del SQTL, principalmente nadolol (0,5-1 mg/kg/día), propranolol (2-4 mg/kg/día), metoprolol (0,5-1 mg/kg/día) y atenolol (0,5-1 mg/kg/día). Sin embargo, el atenolol puede no ser beneficioso en el SQTL; se ha notificado que al menos el 75% de los pacientes que no respondieron al tratamiento con betabloqueantes estaban recibiendo atenolol, aunque este hallazgo puede estar relacionado con el uso de dosis subóptimas.104 La prueba de esfuerzo es útil para establecer la dosis adecuada. La frecuencia cardíaca máxima no debe superar los 130 latidos/min durante el tratamiento.
Bloqueadores del canal de sodio
Las mutaciones del canal de sodio que causan el SQTL3 producen una inactivación defectuosa del canal; el bloqueo del canal de sodio ha demostrado ser útil en estos pacientes. Los estudios realizados con flecainida han documentado mejoras en la frecuencia cardíaca, las alteraciones de la onda T y el intervalo QT.108 También se ha informado de que la mexiletina mejora los marcadores de riesgo electrocardiográfico.63,109,110 Los estudios in vitro con ranolazina han mostrado una disminución de los efectos deletéreos de las mutaciones informadas en humanos.111 Aunque los resultados son alentadores, debe tenerse en cuenta que no hay estudios a largo plazo que evalúen esta terapia, ni se han comunicado resultados de grandes series. Los bloqueadores de los canales de sodio no deben administrarse si no hay un diagnóstico genético confirmado.
Suplementos de potasio y fármacos que aumentan su disponibilidad
Los suplementos de potasio y/o los fármacos ahorradores de potasio, como la espironolactona, acortan el intervalo QTc en el 24% de los casos.112,113 Los fármacos que favorecen la apertura de los canales de potasio, como aprikalim, levcromakalim, nicorandil y pinacidil, han demostrado ser útiles en el tratamiento del SQTL. Los subtipos en los que resultan especialmente beneficiosos son el SQTL1 y el SQTL2.114
Marcapasos y desfibriladores
La estimulación con marcapasos se ha utilizado en pacientes con arritmia dependiente de la pausa.115,116 Los pacientes con SQTL3 suelen beneficiarse más de este tratamiento porque la prevalencia de bradicardia es mayor en este grupo. La estimulación DDD está indicada en pacientes con arritmia dependiente de la pausa o con bloqueo AV 2:1 de alto grado. Las frecuencias programadas por debajo de 70 latidos/min117 no son útiles para prevenir la arritmia ventricular. Se recomienda programar el sensor a respuesta rápida, ya que estos pacientes suelen presentar una aceleración inapropiada de la frecuencia cardíaca en respuesta al ejercicio. Todas las funciones que impliquen la presencia de pausas deben ser desactivadas, como la histéresis y la función nocturna. El PARP (período refractario auricular postventricular) debe ser lo más corto posible. La función de regulación de la frecuencia debe estar activada para evitar la pausa postextrasistólica. Hay que recordar que la sobresensibilidad de la onda T y los fallos de captura también pueden dar lugar a pausas. El uso combinado de un desfibrilador cardioversor implantable (DCI) y de betabloqueantes disminuye sustancialmente la incidencia de muerte súbita.118-120 La indicación de estas medidas es clara en los casos de alto riesgo.121 La programación del dispositivo variará según las necesidades de cada paciente, pero, en general, debe evitarse la administración del tratamiento en eventos asintomáticos y autolimitados; para ello, se indica un tiempo de detección de 15 s. La tormenta arrítmica es una complicación del tratamiento con AID. Cerca del 15% de los pacientes pueden experimentar esta complicación, que se debe, en buena parte, al aumento del tono simpático tras la descarga del DAI.118 Este problema puede gestionarse aumentando la dosis de betabloqueantes. Si esta medida no es útil, debe considerarse la resección de los ganglios de la cadena simpática.
Simpatectomía izquierda
En 1971 se introdujo la gangliectomía simpática como una opción terapéutica útil en estos pacientes.122 En 1991, Schwartz et al123 publicaron la primera serie de 85 pacientes con mala respuesta al tratamiento con betabloqueantes, en los que se realizó una estelectomía izquierda con resultados alentadores: una supervivencia a los 5 años del 94%. En la actualidad, esta opción terapéutica se ofrece a los pacientes de alto riesgo que persisten en el síncope a pesar del tratamiento con betabloqueantes y/o la implantación de un marcapasos, y a los que experimentan descargas frecuentes de su desfibrilador implantado. El procedimiento consiste en la resección de la porción inferior del ganglio estrellado y de los ganglios torácicos izquierdos T2 a T4 de la cadena simpática, ya que la simple estelectomía izquierda no ha demostrado ser suficientemente eficaz. Se ha utilizado la toracoscopia microinvasiva124,125 con buenos resultados. Recientemente se ha publicado la mayor serie de pacientes tratados con este método, que ha mostrado una reducción significativa del número de episodios de síncope o muertes súbitas, así como una tasa de supervivencia a los 5 años del 95%. En los pacientes con síncopes previos, la supervivencia a los 5 años fue del 97%, con una posibilidad de recurrencia del 11%, que, en la mayoría, consistió en un único episodio sincopal. También hubo una reducción significativa del segmento QT tras la simpatectomía izquierda. A pesar de estos resultados favorables, la prevención de la muerte súbita no es completa, pero se ha reducido al 3%. En los pacientes con un CDI que se sometieron a la intervención quirúrgica debido a las múltiples descargas del desfibrilador, el número medio de eventos se redujo de 25 a 0, una reducción del 95%. Se confirmó un efecto beneficioso en el SQTL1. Es probable que los beneficios sean menores en los pacientes con SQTL2, y en el SQTL3 no se ha demostrado su eficacia.126
Ablación
Se ha informado de que la ablación de la extrasístole, que en algunos casos inicia la arritmia ventricular, puede llevarse a cabo con una reducción de la incidencia de episodios.127 Sin embargo, no existen estudios a largo plazo con un número adecuado de pacientes que justifiquen el uso rutinario de esta técnica.
Ver editorial en las páginas 675-82
ABREVIACIONES
AV: auriculoventricular
AID: desfibrilador automático implantable
ECG: electrocardiograma
QTc: QT corregido por la frecuencia cardíaca
ATS: Síndrome de Andersen-Tawil
SQT: síndrome de QT largo
El Dr. Medeiros recibe apoyo económico de CONACyT y FUNSALUD.