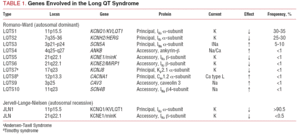
ÚVOD
Syndrom dlouhého QT intervalu (LQTS) je charakterizován závažnou změnou komorové repolarizace, která vede k prodloužení QT intervalu na elektrokardiogramu (EKG). Tento stav predisponuje pacienty k maligní komorové arytmii (torsade de pointes) a náhlé smrti. Klinický a elektrokardiografický popis syndromu dlouhého QT popsali v roce 1957 Anton Jervell a Fred Lange Nielsen,1 kteří publikovali své studie na rodině nepříbuzných rodičů se 6 dětmi. Čtyři z dětí měly vrozenou hluchotu a synkopální epizody a u 3 se projevila náhlá smrt. EKG vyšetření těchto pacientů ukázalo neobvykle dlouhý QT interval. Oba rodiče byli asymptomatičtí, měli normální EKG a neprezentovali žádné problémy se sluchem. V roce 1964 Romano a Ward nezávisle na sobě popsali kardiální syndrom charakterizovaný opakovanými synkopami, rodinnou anamnézou náhlého úmrtí a prodloužením intervalu QT bez neuronální hluchoty.2 Pozdější genetické studie ukázaly, že syndrom popsaný Jervellem a Lange Nielsenem, který je doprovázen vrozenou neuronální hluchotou, odpovídá homozygotním mutacím se závažným fenotypem a vysokým rizikem náhlé smrti. Stav známý jako Romano-Wardův syndrom obecně odpovídá heterozygotním mutacím, pacienti nevykazují změny sluchu a závažnost onemocnění se značně liší. Téměř o půl století později, v roce 1995,3,4 byly popsány hlavní geny spojené s LQTS a onemocnění bylo uznáno jako porucha srdečních iontových kanálů. Jednalo se o první popsanou srdeční kanálopatii, která je doposud pravděpodobně nejrozsáhleji zkoumanou arytmogenní poruchou iontových kanálů. Klinický obraz se značně liší: pacient může být asymptomatický nebo se u něj jako první projev onemocnění mohou objevit opakované synkopy, záchvaty nebo náhlá smrt. Zpočátku byl LQTS považován za vzácný syndrom a v podstatě se jedná o ojedinělé závažné projevy onemocnění. Nicméně výskyt příbuzných mutací se odhaduje na 1/3000-5000 případů,5 32 % asymptomatických nositelů může mít interval QT korigovaný na srdeční frekvenci (QTc) v mezích normy, onemocnění se přenáší na 50 % jejich potomků, jsou náchylnější k rozvoji arytmie ve srovnání s běžnou populací a až 20 % může být symptomatických.6
Syndrom dlouhého QT vykazuje velkou genetickou heterogenitu. U tohoto onemocnění bylo popsáno více než 500 mutací rozmístěných v 10 genech: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 a SCN4B. I přes pokroky v této oblasti se genetickou diagnózu nedaří stanovit u 25-30 % pacientů.7,8 Projevy onemocnění jsou většinou monogenní6 , polygenní nebo složené varianty mají obvykle závažnější fenotyp. Penetrance, tj. počet pacientů, kteří mají mutaci a projevují se daným fenotypem, se pohybuje od 25 % do 90 %.9 Méně často se mohou vyskytovat rozdíly v expresivitě onemocnění, kdy ze stejné mutace vyplývá několik fenotypů. Molekulárně genetické studie vypracované v posledních 11 letech přinesly důležité korelace mezi genotypem a fenotypem, které pomohly nasměrovat léčebný přístup. Kromě toho byla ve studiích zkoumajících časté nesynonymní polymorfismy v této populaci učiněna zajímavá pozorování týkající se individuální náchylnosti k rozvoji arytmie, což je aspekt, který vzbudil značný zájem zejména v oblasti farmakogenomiky.
Klasifikace LONG QT SYNDROMU
Obecné pojmy
Klasifikace LQTS používaná v minulosti byla založena na homozygotní nebo heterozygotní prezentaci onemocnění, která dává vzniknout Jervell-Lange-Nielsenovu syndromu (s hluchotou), resp. syndromu Romano-Ward (bez hluchoty). Současná klasifikace klade důraz na genetické nálezy, jak je znázorněno v tabulce 1. V letech 1995-1996 byly popsány 3 hlavní geny spojené s tímto onemocněním. Tyto geny, které kódují pórotvorné jednotky draslíkových kanálů IKs a IKr a sodíkový kanál Nav1.5, představují téměř 65 % případů. Přestože v následujících letech bylo do seznamu zařazeno dalších sedm genů, tvoří pouze 5 % případů.
Iontové kanály jsou transmembránové proteiny, které transportují ionty buněčnou membránou. Kanály, které se podílejí na LQTS, jsou selektivní nebo specializované na transport jednoho iontu a jsou napěťově závislé, tj. k jejich aktivaci dochází při určitém intracelulárním napětí, které se liší podle podtypu kanálu. Elektrické a kontraktilní jevy, ke kterým dochází v kardiomyocytu, jsou řízeny těmito strukturami. Iontové kanály tvoří makromolekulární komplexy sestávající z hlavní jednotky, která tvoří pór kanálu, a pomocných proteinů, které jej regulují (obr. 1). Dysfunkce kanálů pozorovaná u LQTS se může vyskytovat na těchto dvou místech: na hlavním proteinu nebo na regulačních proteinech (tabulka 1). Zapojení jednotky tvořící póry, známé jako alfa, vytváří tři nejčastější podtypy LQTS: LQTS1 (postihující draslíkový kanál IKs), LQTS2 (postihující draslíkový kanál IKr) a LQTS3 (postihující sodíkový kanál). Protože se jedná o nejčastější podtypy, jsou nejlépe klinicky i geneticky charakterizovány. Korelace fenotypu a genotypu u těchto tří hlavních forem jsou popsány na obrázku 2. V současné době odpovídá Jervell-Lange-Nielsenův syndrom odrůdám LQTS 1 a 5. Pro tyto pacienty je charakteristická vrozená hluchota a složené homozygotní nebo heterozygotní mutace, které ovlivňují proud IKs. Romano-Wardův syndrom zahrnuje odrůdy LQTS 1 až 10 a nezahrnuje hluchotu.
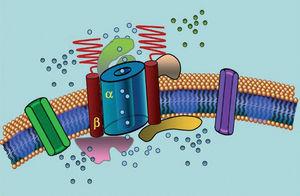
Obr. 1. Syndrom Romano-Ward zahrnuje odrůdy LQTS 1 až 10 a nezahrnuje hluchotu. Schematické znázornění makromolekulárního komplexu. Iontové kanály jsou transmembránové proteiny (α) regulované různými proteiny; jedním z nich je tzv. β podjednotka.
Syndrom dlouhého QT typu 1 (LQTS1)
U pacientů s LQTS1 se obvykle objevují epizody komorové arytmie při cvičení nebo při působení sympatických podnětů (68 %).10 Jako sport vyvolávající arytmii u LQTS1 bylo popsáno plavání.11 Penetrance je u tohoto podtypu téměř 62 %. Vlna T má u těchto pacientů často širokou základnu a velmi prodloužené trvání12,13 (obr. 2). Jedná se o nejčastější podtyp a vysvětluje 30-35 % případů. Postižený gen KvLQT1 (nebo KCNQ1) se nachází na chromozomu 11 (11p15.5) a kóduje α-podjednotku draslíkového kanálu IKs. Akční potenciál je prodloužen snížením odcházejícího proudu K+ během fáze 3 akčního potenciálu.
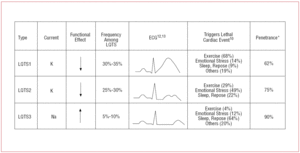
Obrázek 2. Akční potenciál je prodloužen snížením odcházejícího proudu K+ během fáze 3 akčního potenciálu. Korelace genotyp-fenotyp u nejčastějších syndromů dlouhého QT. *Odkazuje na případy, které mají mutaci a projevují se fenotypem.
Syndrom dlouhého QT typu 2 (LQTS2)
U pacientů s LQTS2 se obvykle objevuje komorová arytmie v reakci na emoční stres (49 %) nebo náhlé sluchové podněty (např. budík) a méně často během spánku (22 %) nebo cvičení (29 %).10 Zvláště náchylné jsou ženy v období po porodu.14 Odhadovaná penetrance je 79 %; až 20 % případů tedy může mít nediagnostické EKG. Vlna T u LQTS2 je obvykle nízko amplitudová a bifidní, se zářezy12,13 (obr. 2). Postiženým genem je KCNH2 nebo HERG, který se nachází na chromozomu 7 (7q35-36) a který kóduje α-podjednotku draslíkového kanálu IKr a tvoří 25-30 % případů. Dysfunkce tohoto kanálu snižuje výstupní K+ proud během fáze 3 akčního potenciálu, čímž prodlužuje jeho trvání.
Syndrom dlouhého QT typu 3 (LQTS3)
Pacienti s LQTS3 mají vyšší riziko výskytu maligních arytmií během klidu (spánku) nebo bradykardie.15 Penetrance mutace genu SCN5A je téměř 90%. EKG u LQTS3 obvykle ukazuje opožděnou špičatou vlnu T a umožňuje jasně pozorovat prodloužení úseku ST12,13 (obr. 2). Tito pacienti mají obvykle méně příznaků než pacienti s LQTS1 nebo LQTS2, ale příhody jsou charakteristicky smrtelnější.
Postiženým genem u LQTS3 je SCN5A, který kóduje α-podjednotku sodíkového kanálu Nav1.5 (obr. 1), nacházející se na chromozomu 3 (3p21-24); je příčinou onemocnění v 5-10 % případů. Defektní inaktivace kanálu umožňuje trvalý vstup Na+ během 2. fáze akčního potenciálu, což prodlužuje jeho trvání.
Syndrom dlouhého QT typu 4 (LQTS4)
Typ 4 je vzácnou variantou LQTS, která představuje téměř 1 % případů. Jedná se o atypickou formu, která vyvolává široké spektrum arytmií, včetně katecholaminergní polymorfní komorové tachykardie, fibrilace síní, změn intraventrikulárního vedení, dysfunkce sinusového uzlu a bradykardie6-18; u mnoha pacientů může být navíc QTc v mezích normy. Postiženým genem je ANKB, který se nachází na chromozomu 4 (4q25-27) a který kóduje syntézu ankyrinu-β, strukturálního proteinu, který spojuje membránové proteiny kardiomyocytů s proteiny cytoskeletu. Těmito proteiny jsou Na/K ATPázová pumpa, Na/Ca výměník a inositoltrifosfátový receptor (InsP3R). Mutace způsobující ztrátu funkce ankyrinu-β vedou ke zvýšení intracelulární koncentrace vápníku a ke změnám exprese N/K ATPázy a Na/Ca výměníku. Zvýšená koncentrace vápníku vede k časným a opožděným depolarizacím. Komorové arytmie pozorované u mutací genu pro ankyrin-β jsou tedy způsobeny spontánními depolarizacemi, obvykle v reakci na katecholaminergní stimulaci.
Syndrom dlouhého QT typu 5 (LQTS5)
Typ 5 vzniká při změnách v sekvenci genu KCNE1 umístěného na chromozomu 21 (21q22.1p22.)19 KCNE1 kóduje syntézu β-podjednotky kanálu IKs, známé také jako podjednotka minK, která reguluje kanál IKs. Tento typ tvoří méně než 1 % případů.
Syndrom dlouhého QT typu 6 (LQTS6)
Postiženým genem u typu 6 je KCNE2, který se nachází na chromozomu 21 (21q22.1).20 Tento gen kóduje β-podjednotku draslíkového kanálu, známou také jako podjednotka MiRP1, a reguluje kanál IKr. Méně než 1 % případů je typu 6.
Syndrom dlouhého QT typu 7 neboli Andersenův-Tawilův syndrom (LQTS7)
Dysmorfní nálezy a elektrokardiografické změny pozorované u tohoto syndromu poprvé popsal v roce 1971 Dr. Andersen21 a v roce 1994 se k nim vrátil Dr. Tawil,22 ale genetický/molekulární popis byl uveden až v roce 2001.23 Nyní je toto onemocnění známé jako Andersenův-Tawilův syndrom (ATS) a jedná se o autozomálně dominantní změnu charakterizovanou periodickou paralýzou, abnormálním vývojem kostry, komorovou arytmií typu zahrnující časté komorové extrasystoly a zvláštní náchylností k rozvoji komorové fibrilace, zejména u žen. Mezi změny popsané u ATS patří komorové extrasystoly (41 %), neudržovaná polymorfní komorová tachykardie (23 %), obousměrná komorová tachykardie (68 %) a torsade de pointes (3 %).24 Mezi pozorované dysmorfické znaky patří malý vzrůst, skolióza, klinodaktylie, hypertelorismus, nízká implantace uší, mikrognatie a široké čelo. Projevy onemocnění jsou různé, což komplikuje včasnou diagnostiku.23,25 Mutace v genu KCNJ2 umístěném na 17. chromozomu (17q23), který kóduje syntézu rektifikačního draslíkového kanálu Kir 2.1, tvoří 70 % případů. Tento kanál se podílí na 4. fázi akčního potenciálu. Někteří autoři zpochybňují zařazení tohoto genu do kauzální skupiny LQTS, protože interval QTc je u tohoto syndromu prodloužen jen mírně nebo dokonce normálně, ale vlna U je obvykle výrazná, což vedlo k nadhodnocení intervalu QT. Čtenář se dozví, že někteří autoři se domnívají, že mutace KCNJ2 generují ATS1, a nikoli LQTS7.24
Syndrom dlouhého QT typu 8 (LQTS8)
Typ 8 vzniká na základě mutací v genu CACNA1 umístěném na chromozomu 12 (12p13.3), který kóduje vápníkový kanál typu L Cav1.2. Způsobuje Timothyho syndrom26 , který se vyznačuje srdečními malformacemi, intermitentním imunologickým deficitem, hypoglykémií, kognitivními změnami včetně autismu, interdigitální fúzí a prodlouženým QT, což vede k srdeční arytmii a náhlé smrti27. Méně než 0,5 % případů je typu 8.
Syndrom dlouhého QT typu 9 (LQTS9)
Tato varianta LQTS se vyvíjí na základě mutací v genu CAV3, který se nachází na chromozomu 3 (3p25) a který kóduje syntézu kaveolinu 3. Syndrom dlouhého QT typu 9 (LQTS9) se vyvíjí na základě mutací v genu CAV3, který se nachází na chromozomu 3 (3p25). Kaveol je invaginace plazmatické membrány, která se podílí na endocytóze, homeostáze lipidů a přenosu signálu. Důležitou součástí této struktury je kaveolin, který má 3 známé podtypy; podtyp 3 je specifický pro kosterní a srdeční sval. Některé iontové kanály jsou umístěny v kaveole, včetně srdeční izoformy sodíkového kanálu Nav1.5. V poslední době bylo popsáno několik mutací tohoto proteinu. Ty mění biofyzikální vlastnosti sodíkového kanálu Nav1.5 in vitro a vytvářejí fenotyp podobný tomu, který je pozorován u LQTS3.28 Této příčině se přisuzuje méně než 1 % případů.
Syndrom dlouhého QT typu 10 (LQTS10)
Typ 10 byl popsán u velmi závažného případu s QTc >600 ms, fetální bradykardií a atrioventrikulární (AV) blokádou 2:1. Je důsledkem mutací v genu SCN4B, který se nachází na chromozomu 11 (11q23) a kóduje β4-podjednotku sodíkového kanálu. Byly popsány čtyři různé podtypy podjednotek β, které interagují a regulují různé izoformy sodíkového kanálu, nicméně s arytmogenezí byl dosud spojován pouze podtyp 4.29 Výskyt mutací tohoto genu nebyl zkoumán, ale odhaduje se na
Mutace Jervell-Lange-Nielsenovy variety
Tato těžká forma LQTS je způsobena homozygotními30 nebo složenými heterozygotními mutacemi genů KCNQ1, a/nebo KCNE1, které kódují proud IKs; tj. velmi těžká varieta formy LQTS1 nebo LQTS5. Tento stav je charakteristicky spojen s vrozenou hluchotou. Pacienti mají obvykle QTc>500 ms a opakované synkopy a jsou vystaveni vysokému riziku náhlé smrti. Rodiče pacientů s touto variantou jsou obvykle heterozygoti a mají méně závažné onemocnění nebo nevykazují žádné příznaky.31
DIAGNÓZA SYNDROMU DLOUHÉHO QT
Schwartzovo skóre
V roce 1985 Schwartz a spol.32 publikovali kritéria pro diagnostiku LQTS, která byla upravena v roce 1993 a obsahují důležité pokyny pro počáteční hodnocení potenciálních případů. Tento systém používá skóre od 1 do 9 na základě rodinné anamnézy a klinických a elektrokardiografických nálezů. Pravděpodobnost onemocnění je nízká při skóre ≥1, střední při 2-3 a vysoká při ≥4 (tabulka 2).

Prenatální diagnostika syndromu dlouhého QT
Fetální bradykardie může být jedním z prvních klinických projevů LQTS. Retrospektivní série ukázaly, že až 70 % pacientů s diagnostikovaným LQTS v dětství má v anamnéze bradykardii, obvykle doprovázenou hydropsem plodu.33 Hodnocení srdeční repolarizace plodu mezi 14. a 39. týdnem je užitečné pro časnou diagnostiku LQTS.34
Gonadální mozaika pro LQTS byla spojena s opakovanými ztrátami plodu během třetího trimestru těhotenství.35 Při velkém podezření na onemocnění může být pro stanovení diagnózy užitečná amniocentéza po 16. týdnu těhotenství, které lze snadno dosáhnout, pokud je známo, že jeden z rodičů je nositelem specifické mutace.36
STUDIE PACIENTA SE SYNDROMEM DLOUHÉHO QT
Klinická anamnéza
Pro diagnózu i stratifikaci rizika LQTS má zásadní význam rodinná a/nebo osobní anamnéza náhlé smrti. Kromě toho mohou precipitující faktory a kontext synkopy indikovat podtyp LQTS. Při počátečním hodnocení podezřelého případu je třeba vyloučit užívání léků, které mohou prodloužit interval QT.
Interval QT:
Interval QT by se měl přednostně měřit ve svodech II nebo V5,37 kde se prokázalo, že má větší výpovědní hodnotu.38 Tento interval udává dobu trvání komorové repolarizace a měří se od začátku vlny Q do konce vlny T. Obvykle se ke korekci trvání intervalu podle srdeční frekvence používá vzorec navržený Bazettem39 (QTc=QT/√RR, vyjádřeno v sekundách). Ačkoli se měření intervalu QT zdá být jednoduché, v multicentrické studii provedené Viskinem et al40 vědělo, jak jej správně změřit, méně než 40 % lékařů jiných než kardiologů, méně než 50 % kardiologů a více než 80 % specialistů na arytmie. Je vhodné, aby lékaři prováděli manuální měření a nedůvěřovali automatickým měřením, která mohou být užitečná pro jiné intervaly, ale při výpočtu QT intervalu jsou nepřesná. QT interval je dynamický interval a jeho normální hranice závisí na několika faktorech. Ačkoli je interval QTc é440 ms u mužů a é460 ms u žen považován za abnormální, lze v tomto rozmezí nalézt nositele mutací i zdravé jedince (obr. 3). V rodinách s LQTS1 Vincent et al41 prokázali, že žádný z případů s pozitivním genotypem neměl QTc470 ms. Monnig et al38 nedávno ukázali, že QTc>440 ms postačuje k odhalení pacientů s mutacemi spojenými s LQTS, QTc>470 ms je užitečné k identifikaci pacientů s rizikem rozvoje příznaků a QTc>500 ms se nachází u symptomatických pacientů podstupujících léčbu.
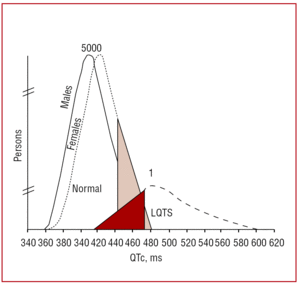
Obrázek 3. Model znázorňující rozložení QT intervalu korigovaného na srdeční frekvenci (QTc) u pacientů s mutacemi v KVLQT1, HERG nebo SCN5A a jejich nepostižených rodinných příslušníků. Křivka vlevo popisuje distribuci nepostižených členů a křivka vpravo postižené členy.
Další elektrokardiografické změny spojené se syndromem dlouhého QT
U pacientů s LQTS se mohou vyskytovat četné změny vlny T: mimo jiné alterace polarity, změny amplitudy, vrubování a bifázický vzhled.42 Alternans vlny T (obr. 4A) je definován jako kolísání amplitudy, morfologie a polarity vlny T v sinusovém rytmu po jednotlivých taktech, bez kolísání komplexu QRS. Je ukazatelem elektrické nestability,43 odráží regionální rozptyl komorové repolarizace a příležitostně předchází komorové fibrilaci.44
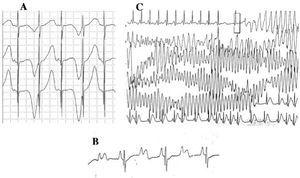
Obrázek 4. Vlna střídavých vln v komoře. Elektrokardiografické změny u syndromu dlouhého QT. A: Elektrické alternace vlny T. B: atrioventrikulární blokáda 2:1. C: samostatně omezená torsade de pointes.
U pacientů s LQTS mohou progredovat známky dysfunkce sinusového uzlu, bradykardie a/nebo pauzy.45 U podtypů LQTS1 a LQTS3, zejména u druhého z nich, se často vyskytuje sinusová bradykardie,46 zatímco LQTS4 je spojen s dysfunkcí sinusového uzlu.18
Od desetiletí 1970-1980 je pozorována koexistence defektů AV vedení s LQTS47 (obr. 4B). AV blokáda dva ku jedné je vzácným projevem se špatnou prognózou, který může být přítomen již od fetálního stadia v podobě perzistující bradykardie. Incidence této abnormality byla zaznamenána ve 4-5 %48 a je spojena s vysokou mortalitou navzdory léčbě betablokátory a/nebo kardiostimulátory.49,50 Tento jev lze vysvětlit dlouhým trváním akčního potenciálu. Při prodloužení refrakterní periody komor je následující impuls sinusové aktivity blokován, protože se ke komorám dostane ještě v době, kdy jsou v refrakterní periodě. Zdá se, že k této změně dochází výhradně u LQTS, protože refrakterní perioda komor je delší než perioda AV převodního systému.51 Sklon QRS komplexu je obvykle strmý a blok byl lokalizován v infraHis oblasti,46,51,52 ale místo bloku může záviset na genotypu. Dosud byly s blokem 2:1 u LQTS spojeny 4 geny: HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 a SCN4B (LQTS10).55
Komorová arytmie charakteristická pro LQTS se nazývá torsade de pointes (obr. 4C). Projevuje se při prodloužení intervalu QT bez ohledu na etiologii. Jedná se o polymorfní komorovou tachykardii způsobenou reentry, která je elektrokardiograficky charakterizována kontinuálním stáčením osy QRS kolem pomyslné čáry. Běžně jí předchází pauza následovaná extrasystolou (krátký-dlouhý-krátký RR interval), jak je znázorněno na obrázku.56-58 Může vyvrcholit komorovou fibrilací a náhlou smrtí. Pokud k tomu nedojde, může pacient zažít pouze synkopu, a pokud je epizoda krátká, může zůstat neodhalena.
Holterova
Holterova studie poskytuje úplné, dynamické posouzení QT intervalu. Občas jsou zaznamenány spontánní epizody asymptomatické komorové arytmie, stejně jako epizody dysfunkce sinusového uzlu nebo AV blokády.
Zátěžový test při zátěži
Pacienti s LQTS nemohou dosáhnout maximální očekávané tepové frekvence vypočtené podle věku. Kromě toho může při zátěži interval QT vykazovat paradoxní chování tím, že se spíše prodlužuje, než zkracuje.59,60 Elektrokardiografický obraz při zátěžovém testu se bude lišit v závislosti na typu LQTS. Pacienti s LQTS1 kromě toho, že nedosáhnou maximální vypočtené tepové frekvence pro svůj věk, často vykazují prodloužení intervalu QT, zatímco pacienti s LQTS2 mohou dosáhnout očekávané tepové frekvence a vykazovat pouze mírné prodloužení intervalu QT nebo žádné.61,62 Pacienti s LQTS3 mají obecně fyziologickou odpověď na zátěž, tj. normální zkrácení intervalu QT.63 Zátěžové testy mohou být užitečné také pro posouzení odpovědi na léčbu a pro stratifikaci rizika u asymptomatických případů nebo v případě pochybností o příhodách vedoucích k arytmii.
Genetický screening
V posledních letech byly genetické studie u LQTS omezeny na výzkumné laboratoře. Nicméně informace získané z těchto snah jsou velmi užitečné pro léčbu pacientů, zejména vysoce rizikových případů. Snad hlavní uplatnění screeningu je v genetickém poradenství, ale má také důležité důsledky v léčbě, kterou lze orientovat podle postiženého kanálu. Přesná lokalizace dané mutace může poskytnout další informace týkající se vývoje rizika. Pacienti s mutacemi v transmembránové oblasti KCNQ1 (IKS) mají větší pravděpodobnost výskytu arytmických příhod než pacienti s mutacemi v C-koncové oblasti64; totéž platí pro pacienty s mutacemi v pórové oblasti KCNH2 nebo HERG65 ve srovnání s pacienty s mutacemi v N- nebo C-koncové oblasti.66
Počáteční screening se snad může omezit na geny KCNQ1, HERG a SCN5A, které poskytují možnost narazit na mutace v 65 % případů. Pokud jsou získané výsledky negativní, lze screening rozšířit na geny KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 a SCN4B, což zvýší možnost pozitivních výsledků o 5-10 %.
Posmrtný genetický screening
Zajímavé je, že mutace genů vedoucí k LQTS byly nalezeny u dětí, u nichž došlo k náhlé smrti, a v nevysvětlitelných případech náhlé smrti u mladých dospělých.
Postmortální genetické studie pacientů s náhlým úmrtím a negativní pitvou prokázaly mutace vedoucí k LQTS v různém procentu67-69: téměř 10 % u dětí a 35 % u mladých dospělých.70-72 Na základě těchto výsledků bylo navrženo rutinní vyšetření EKG u všech novorozenců.73,74
Postmortální genetická studie, v literatuře známá také jako „molekulární pitva“, má kromě právních důsledků i důležité důsledky v rodinách, které mohou být postiženy, aniž by o tom věděly.75
Regulační polymorfismy
V populaci s LQTS bylo popsáno několik často se vyskytujících polymorfismů, které se vyskytují téměř ve všech genech spojených s tímto onemocněním. Ačkoli tyto změny zřejmě nejsou patogenní, některé z nich mohou mít následující účinky75-78:
1. Vlivem těchto změn může dojít k poruchám metabolismu. Vytvářejí individuální náchylnost k rozvoji arytmie.
2. Vlivem těchto faktorů se může vyvinout arytmie. Zvýhodňují patogenní vliv jiné nesynonymní změny.
3. Snižují patogenní vliv jiné nesynonymní změny.
To je případ polymorfismu K897T v genu KCNH2 (HERG), který se vyskytuje až u 15 % populace a je nejen spojen s náchylností k některým lékům,79 ale také zvýhodňuje patogenní vliv mutací ve stejném genu.78 Dalším příkladem je polymorfismus S1103Y v genu SCN5A, který se vyskytuje především u černochů, má téměř 13% výskyt a je spojen se zvýšeným rizikem náhlé smrti v dětství.80
Zajímavé je, že v produktu genu SCN5A (který u lidí kóduje izoformu sodíkového kanálu Nav1.5) byla popsána dvě alternativní místa zpracování generující dva typy sodíkových kanálů: jedno s 2016 aminokyselinami obsahujícími glutamin v pozici 1077 (Q1077) a druhé s 2015 aminokyselinami bez glutaminu (Q1077del). Transkripty těchto alternativních zpracování jsou ve stejném lidském srdci přítomny v poměru 2:1 a několik častých polymorfismů bude mít různý vliv na fungování kanálu v závislosti na tom, zda se jedná o kontext Q1077 nebo Q1077del. To bylo původně prokázáno u polymorfismu H558R SCN5A, který se vyskytuje až u 30 % populace. Když byl H558R vyjádřen v kontextu Q1077, bylo pozorováno hluboké snížení iontového proudu.81 Podobný účinek byl dokumentován u polymorfismu S524Y82. Tyto nálezy poskytly faktory vysvětlující různou závažnost onemocnění i různé fenotypy téže mutace pozorované v některých rodinách.77
Farmakologické vyšetření adrenalinem
Farmakologické vyšetření nízkou dávkou adrenalinu je bezpečnou a užitečnou možností, jak demaskovat podezřelé případy LQTS s hraničním QTc. Je obzvláště účinné při odhalování asymptomatických forem LQTS1, se senzitivitou 92,5 %, specificitou 86 %, pozitivní prediktivní hodnotou 76 % a negativní prediktivní hodnotou 96 %. Může být také užitečný při diagnostice LQTS2, s nižší senzitivitou a specificitou. Není užitečná pro LQTS3 nebo jiné formy LQTS. Za normálních podmínek vyvolává sympatická stimulace fosforylaci draslíkového kanálu IKs, což optimalizuje jeho funkci a vede ke zkrácení akčního potenciálu. U pacientů s LQTS, zejména typu 1, je pozorována paradoxní reakce na podání nízké dávky adrenalinu (0,025-0,2 µg/kg/min), která prodlužuje interval QT na více než 30 ms83-86.
Prodloužení intervalu QT a léky způsobené TORSADE DE POINTES
Velké množství léků používaných v různých lékařských specializacích může způsobit iatrogenní prodloužení intervalu QT. Některé léky byly kvůli tomuto nežádoucímu účinku staženy z trhu (např. astemizol a cisaprid, mimo jiné; více informací naleznete na www.qtdrugs.org).87,88
Komorová arytmie sekundárně způsobená neantiarytmickými léky se vyskytuje u méně než jednoho z 10 000 až 100 000 exponovaných subjektů. Vzhledem k tomu, že klinické studie zahrnují 2000 až 3000 subjektů, tato nežádoucí a fatální nežádoucí příhoda by snadno unikla odhalení během klinické fáze vývoje léku.89 Tento bod vyvolal obrovský zájem o aspekty týkající se bezpečnosti při studiu a vývoji nových léků.
Faktory související s individuální náchylností zahrnují ženské pohlaví, hypokalcemii, hypomagnezemii, bradykardii, srdeční selhání, postkardioverzi, fibrilaci síní, hypertrofii levé komory, nezjištěný LQTS, predisponující polymorfismy a vysoké sérové koncentrace predisponujících léků.90
Kanál, který typicky interaguje s léky, je IKr, kódovaný genem KCNH2(HERG), a to kvůli své molekulární struktuře. Ostatní draslíkové kanály mají 2 prolinové zbytky skloněné směrem k póru kanálu, což zmenšuje jeho lumen. Naproti tomu IKr tyto zbytky postrádá, vytváří se větší předsíň póru a usnadňuje se expozice velkým molekulám. Kromě toho má 2 aromatická rezidua (tyrosin a fenylalanin), která zvýhodňují vazbu s aromatickými molekulami přítomnými v několika lécích schopných blokovat kanál.91
Jak bylo uvedeno výše, penetrance LQTS je neúplná a u některých asymptomatických nositelů mutací se může po podání některého z těchto léků projevit maligní arytmie. Kromě toho polymorfismy považované za časté v populaci propůjčují individuální náchylnost ke vzniku torsade de pointes při užívání některých léků. To je případ polymorfismu R1047L, druhého nejčastějšího v KCNH2, který byl spojen s rozvojem torsade de pointes při užívání léku dofetilid.92 U zdravých osob bylo popsáno nejméně 20 polymorfismů genu KCNH2 a jejich vliv na individuální náchylnost k rozvoji arytmie související s léky je třeba ještě určit.93 Polymorfismy, které propůjčují náchylnost k rozvoji komorové arytmie, byly dokumentovány také u sodíkového kanálu Na1.5. Jedná se o polymorfismus H558R, který je přítomen až u 30 % populace, nebo S1103Y, který je častý u černochů80,81,90,94,95 ; jejich vliv na náchylnost k lékům nebyl zkoumán.
SYNDROM DLOUHÉHO QT ČASU A TĚHOTENSTVÍ
Genetické poradenství je u LQTS důležité, ale obecně platí, že u žen, které jsou jeho nositelkami, není těhotenství kontraindikováno, i když každý případ je jiný a měl by být posuzován individuálně v příslušných souvislostech.
Bylo zjištěno, že riziko výskytu maligní komorové arytmie se s těhotenstvím snižuje. Naopak větší náchylnost k přítomnosti maligní arytmie byla zaznamenána během prvních 9 měsíců po porodu, zejména u pacientek s LQTS2. Toto riziko se výrazně snižuje při léčbě beta-blokátory.96
STRATIFIKACE RIZIKA
Vývoj LQTS se liší a je ovlivněn délkou intervalu QTc, faktory prostředí, věkem, genotypem a odpovědí na léčbu.97,98 Komorová arytmie se častěji vyskytuje u LQTS1 a LQTS2, ale je závažnější u LQTS3.99 Jak bylo uvedeno výše, ženy jsou obzvláště náchylné k maligní arytmii v poporodním období.14
Syndrom dlouhého QT by měl být považován za vysoce rizikový, pokud je spojen s následujícím:
1. Syndrom dlouhého QT je spojen s maligní arytmií. Vrozená hluchota (Jervell-Lange-Nielsenův syndrom).
2. Vrozená hluchota (Jervell-Lange-Nielsenův syndrom). Opakované synkopy v důsledku maligní komorové tachyarytmie.
3. Náhlá smrt v rodinné anamnéze.
4. Náhlá smrt v rodinné anamnéze. QTc>500 ms.
5. Atrioventrikulární blokáda 2:1.
6. Kardiovaskulární blokáda. Elektrický alternans vlny T.
7. Genotyp LQTS3.
Studie Prioriho et al97 provedená u 647 pacientů ukázala, že pravděpodobnost výskytu závažné příhody (synkopa, srdeční zástava, náhlá smrt) před 40. rokem věku je vysoká (>50 %), pokud je QTc >500 ms u LQTS1, LQTS2 a u mužů s LQTS3. Nedávno byla zveřejněna analýza mezinárodního registru LQTS. Riziko náhlé smrti bylo analyzováno u 2772 adolescentů s tímto onemocněním a byly identifikovány 3 faktory spojené s vyšším rizikem v této populaci: QTc>530 ms, anamnéza synkopy v posledních 10 letech a pohlaví; 10-12letí chlapci měli vyšší riziko než dívky, ale ve věkovém rozmezí 13-20 let bylo riziko srovnatelné.100
LÉČBA
Symptomatičtí pacienti, kteří nejsou léčeni, mají roční úmrtnost 20 % a 10letou úmrtnost 50 % po první příhodě komorové arytmie. Ačkoli je zřejmé, že léčba by měla být zavedena při výskytu symptomů, přístup, který je třeba použít u asymptomatických pacientů, je stále předmětem diskusí. Bylo zdokumentováno, že srdeční zástava může být prvním projevem onemocnění u 9 % pacientů48 a že u 12 % asymptomatických pacientů se objeví příznaky a může dojít k náhlé smrti. U všech pacientů s LQTS by měla být zahájena počáteční léčba beta-blokátory. Doporučuje se omezení fyzické zátěže, ale užitečným podkladem pro rozhodování jsou klinické a elektrokardiografické rizikové markery. Je důležité informovat pacienty o riziku užívání několika léků, které mohou prodloužit QT interval a zvýhodnit vznik komorové arytmie, jak je uvedeno výše. Genetická diagnóza, kromě toho, že umožňuje vhodné rodinné poradenství v souvislosti s onemocněním, je pomůckou pro posouzení prognózy a orientaci specifické léčby.
Beta-blokátory
Beta-blokátory jsou lékem první volby u LQTS a všichni pacienti by je měli dostávat jako počáteční léčbu.101 Zajišťují snížení rizika kardiovaskulárních příhod až o 64 %100 a jsou zvláště účinné u pacientů s mutací kanálů IKs (LQTS1)102 , které jsou do značné míry regulovány sympatickým systémem. Beta-blokátory nemění interval QT, ale jeho rozptyl.103 Přestože tyto léky snižují výskyt příhod,104,105 bylo prokázáno, že 10 % pacientů s LQTS1, 23 % s LQTS2 a 32 % s LQTS3 bude mít kardiovaskulární příznaky i přes léčbu.106 Zdá se, že zejména pacienti s LQTS3 nezískávají významné výhody; ve skutečnosti by se tato skupina léků měla u těchto pacientů používat s opatrností, protože epizody komorové arytmie u LQTS3 jsou častější při nízké srdeční frekvenci. Obecně lze říci, že 32 % symptomatických pacientů bude mít během prvních 5 let před zahájením léčby betablokátory opakované příznaky a 14 % pacientů zachráněných z epizody náhlé smrti bude mít během 5 let další podobnou příhodu, pokud budou dostávat pouze tuto léčbu.107 V léčbě LQTS se používá několik beta-blokátorů, především nadolol (0,5-1 mg/kg/den), propranolol (2-4 mg/kg/den), metoprolol (0,5-1 mg/kg/den) a atenolol (0,5-1 mg/kg/den). Atenolol však nemusí být u LQTS prospěšný; bylo oznámeno, že nejméně 75 % pacientů, kteří nereagovali na léčbu beta-blokátory, dostávalo atenolol, ačkoli toto zjištění může souviset s používáním suboptimálních dávek.104 Ke stanovení vhodné dávky je užitečné provádět zátěžové testy. Maximální tepová frekvence by během léčby neměla překročit 130 tepů/min.
Blokátory sodíkového kanálu
Mutace sodíkového kanálu, které způsobují LQTS3, způsobují defektní inaktivaci kanálu; u těchto pacientů se osvědčila blokáda sodíkového kanálu. Studie provedené s flekainidem dokumentovaly zlepšení srdeční frekvence, změn vlny T a intervalu QT.108 Bylo rovněž zaznamenáno zlepšení elektrokardiografických rizikových markerů mexiletinem.63,109,110 Studie in vitro s ranolazinem prokázaly snížení škodlivých účinků mutací zaznamenaných u lidí.111 Ačkoli jsou výsledky povzbudivé, je třeba mít na paměti, že neexistují žádné dlouhodobé studie hodnotící tuto léčbu a nejsou hlášeny žádné výsledky z velkých sérií. Blokátory sodíkových kanálů by se neměly podávat, pokud není potvrzena genetická diagnóza.
Doplňky draslíku a léky zvyšující jeho dostupnost
Doplňky draslíku a/nebo léky šetřící draslík, jako je spironolakton, zkracují interval QTc ve 24 % případů.112,113 Léky, které podporují otevření draslíkových kanálů, jako jsou aprikalim, levcromakalim, nikorandil a pinacidil, se ukázaly jako užitečné při léčbě LQTS. Podtypy, u kterých jsou zvláště přínosné, jsou LQTS1 a LQTS2.114
Kardiostimulátory a defibrilátory
U pacientů s arytmií závislou na pauze se používá stimulace kardiostimulátoru.115,116 Pacienti s LQTS3 mají z této léčby obvykle větší prospěch, protože prevalence bradykardie je u této skupiny vyšší. DDD stimulace je indikována u pacientů s arytmií závislou na pauze nebo AV blokádou vysokého stupně 2:1. Frekvence naprogramované pod 70 tepů/min117 nejsou pro prevenci komorové arytmie užitečné. Doporučuje se naprogramovat snímač na rychlou odezvu, protože u těchto pacientů obvykle dochází k nepřiměřenému zrychlení srdeční frekvence v reakci na cvičení. Všechny funkce, které předpokládají přítomnost pauz, by měly být vypnuty, například hystereze a noční funkce. PARP (postventrikulární atriální refrakterní perioda) by měla být co nejkratší. Funkce regulace frekvence by měla být zapnuta, aby se zabránilo vzniku postxtrasystolické pauzy. Je třeba mít na paměti, že přecitlivělost vlny T a selhání záchytu mohou rovněž vést ke vzniku pauz. Kombinované použití implantabilního kardioverteru-defibrilátoru (ICD) a beta-blokátorů podstatně snižuje výskyt náhlé smrti.118-120 Indikace těchto opatření je jasná ve vysoce rizikových případech.121 Programování přístroje se bude lišit podle potřeb konkrétního pacienta, ale obecně je třeba se vyhnout podávání léčby u asymptomatických, samovolně probíhajících příhod; za tímto účelem je indikována doba detekce 15 sekund. Arytmická bouře je komplikací léčby AID. Téměř u 15 % pacientů se může vyskytnout tato komplikace, která je z dobré části způsobena zvýšeným tonusem sympatiku po šoku ICD.118 Tento problém lze zvládnout zvýšením dávky betablokátoru. Pokud toto opatření nepomůže, je třeba zvážit resekci ganglií sympatického řetězce.122
Levostranná sympatektomie
V roce 1971 byla sympatická gangliektomie představena jako užitečná terapeutická možnost u těchto pacientů.122 V roce 1991 Schwartz a spol.123 publikovali první sérii 85 pacientů se špatnou odpovědí na léčbu beta-blokátory, u kterých byla provedena levostranná stelektomie s povzbudivými výsledky: pětileté přežití 94 %. V současné době se tato terapeutická možnost nabízí vysoce rizikovým pacientům, u nichž přetrvává synkopa navzdory léčbě betablokátorem a/nebo implantaci kardiostimulátoru, a těm, u nichž dochází k častým výbojům z implantovaného defibrilátoru. Zákrok spočívá v resekci dolní části hvězdicového ganglia a T2 až T4 levého hrudního ganglia sympatického řetězce, protože prostá levostranná stellektomie se neukázala jako dostatečně účinná. S dobrými výsledky byla použita mikroinvazivní torakoskopie124,125 . Nedávno byla popsána největší série pacientů léčených touto metodou, která prokázala významné snížení počtu epizod synkopy nebo náhlých úmrtí a také pětileté přežití 95 %. U pacientů s předchozí synkopou bylo 5leté přežití 97 % s 11% možností recidivy, která ve většině případů spočívala v jedné synkopální příhodě. Po levostranné sympatektomii došlo také k významnému zkrácení QT segmentu. Navzdory těmto příznivým výsledkům není prevence náhlé smrti úplná, ale byla snížena na 3 %. U pacientů s ICD, kteří podstoupili operaci z důvodu opakovaných výbojů defibrilátoru, se průměrný počet příhod snížil z 25 na 0, což představuje 95% snížení. Příznivý účinek byl potvrzen u LQTS1. U pacientů s LQTS2 je přínos pravděpodobně menší a u LQTS3 nebyla její účinnost prokázána.126
Ablace
Uvádí se, že ablace extrasystoly, která v některých případech iniciuje komorovou arytmii, může být provedena se snížením výskytu příhod.127 Neexistují však dlouhodobé studie s odpovídajícím počtem pacientů, které by ospravedlnily rutinní používání této techniky.
Viz úvodník na stranách 675-82
Zkratky
AV: atrioventrikulární
AID: automatický implantabilní defibrilátor
ECG: elektrokardiogram
QTc: QT korigovaný srdeční frekvencí
ATS: Andersen-Tawilův syndrom
LQTS: syndrom dlouhého QT intervalu
Dr. Medeiros získává ekonomickou podporu od CONACyT a FUNSALUD.