EINLEITUNG
Das lange QT-Syndrom (LQTS) ist durch eine stark veränderte ventrikuläre Repolarisation gekennzeichnet, die zu einer Verlängerung des QT-Intervalls im Elektrokardiogramm (EKG) führt. Diese Erkrankung prädisponiert die Patienten für bösartige ventrikuläre Arrhythmien (Torsade de pointes) und plötzlichen Tod. Die klinische und elektrokardiografische Beschreibung des langen QT-Syndroms wurde 1957 von Anton Jervell und Fred Lange Nielsen1 veröffentlicht, die ihre Studien an einer Familie nicht blutsverwandter Eltern mit sechs Kindern publizierten. Vier der Kinder wiesen angeborene Taubheit und synkopale Episoden auf, und bei 3 Kindern trat ein plötzlicher Tod ein. Die EKG-Untersuchung dieser Patienten zeigte ein ungewöhnlich langes QT-Intervall. Beide Eltern waren asymptomatisch, hatten ein normales EKG und wiesen keine Hörprobleme auf. 1964 berichteten Romano und Ward unabhängig voneinander über ein kardiales Syndrom, das durch wiederkehrende Synkopen, einen plötzlichen Tod in der Familienanamnese und eine Verlängerung des QT-Intervalls ohne neuronale Taubheit gekennzeichnet ist.2 Spätere genetische Studien zeigten, dass das von Jervell und Lange Nielsen beschriebene Syndrom, das mit einer angeborenen neuronalen Taubheit einhergeht, homozygoten Mutationen mit einem schweren Phänotyp und einem hohen Risiko eines plötzlichen Todes entspricht. Das als Romano-Ward-Syndrom bekannte Krankheitsbild entspricht im Allgemeinen heterozygoten Mutationen, die Patienten weisen keine Hörveränderungen auf, und der Schweregrad der Erkrankung ist sehr unterschiedlich. Fast ein halbes Jahrhundert später, im Jahr 1995,3,4 wurden die wichtigsten mit LQTS assoziierten Gene beschrieben, und die Krankheit wurde als kardiale Ionenkanalstörung anerkannt. Es war die erste kardiale Kanalopathie, die beschrieben wurde, und ist vielleicht die bis heute am umfassendsten untersuchte arrhythmogene Ionenkanalstörung. Das klinische Bild ist sehr unterschiedlich: Die Patienten können asymptomatisch sein oder wiederkehrende Synkopen, Krampfanfälle oder plötzliche Todesfälle als erste Manifestation der Krankheit aufweisen. Ursprünglich galt LQTS als seltenes Syndrom, und tatsächlich tritt die schwere Form der Krankheit nur sporadisch auf. Dennoch wird die Inzidenz der entsprechenden Mutationen auf 1/3000-5000 Fälle geschätzt,5 32 % der asymptomatischen Träger können ein herzfrequenzkorrigiertes QT-Intervall (QTc) innerhalb normaler Grenzen haben, die Krankheit wird an 50 % ihrer Nachkommen weitergegeben, sie sind im Vergleich zur Allgemeinbevölkerung anfälliger für Herzrhythmusstörungen, und bis zu 20 % können symptomatisch werden.6
Das lange QT-Syndrom weist eine große genetische Heterogenität auf. Mehr als 500 Mutationen, verteilt auf 10 Gene, wurden bei dieser Erkrankung beschrieben: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 und SCN4B. Trotz der Fortschritte auf diesem Gebiet kann bei 25-30 % der Patienten keine genetische Diagnose gestellt werden.7,8 Die Krankheit ist hauptsächlich monogen6; polygene oder zusammengesetzte Varianten haben in der Regel einen schwereren Phänotyp. Die Penetranz, d. h. die Zahl der Patienten, die die Mutation aufweisen und den Phänotyp manifestieren, liegt zwischen 25 % und 90 %.9 Seltener kommt es zu Variationen in der Expressivität der Krankheit, wobei mehrere Phänotypen aus derselben Mutation resultieren können. Molekulargenetische Studien, die in den letzten 11 Jahren durchgeführt wurden, haben wichtige Korrelationen zwischen Genotyp und Phänotyp ergeben, die bei der Festlegung des Behandlungsansatzes hilfreich waren. Darüber hinaus wurden in Studien, die die häufigen nicht-synonymen Polymorphismen in dieser Population untersuchten, interessante Beobachtungen zur individuellen Anfälligkeit für die Entwicklung von Herzrhythmusstörungen gemacht, ein Aspekt, der insbesondere im Bereich der Pharmakogenomik großes Interesse geweckt hat.
Klassifizierung des LONG-QT-SYNDROMS
Allgemeine Konzepte
Die in der Vergangenheit verwendete LQTS-Klassifizierung basierte auf der homozygoten oder heterozygoten Ausprägung der Krankheit, die zum Jervell-Lange-Nielsen-Syndrom (mit Taubheit) bzw. zum Romano-Ward-Syndrom (ohne Taubheit) führt. Bei der vorliegenden Klassifizierung wird der Schwerpunkt auf den genetischen Befund gelegt, wie in Tabelle 1 dargestellt ist. Die 3 wichtigsten Gene, die mit der Krankheit in Verbindung gebracht werden, wurden 1995-1996 beschrieben. Diese Gene, die für die porenbildenden Einheiten der Kaliumkanäle IKs und IKr sowie für den Natriumkanal Nav1.5 kodieren, sind für fast 65 % der Fälle verantwortlich. Obwohl in den folgenden Jahren sieben weitere Gene in die Liste aufgenommen wurden, machen sie nur 5 % der Fälle aus.
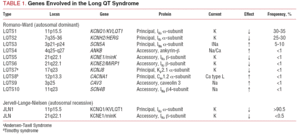
Ionenkanäle sind Transmembranproteine, die Ionen durch die Zellmembran transportieren. Die in LQTS involvierten Kanäle sind selektiv oder auf den Transport eines einzelnen Ions spezialisiert und spannungsabhängig, d. h. ihre Aktivierung erfolgt bei einer bestimmten intrazellulären Spannung, die je nach Kanalsubtyp variiert. Die elektrischen und kontraktilen Phänomene, die im Kardiomyozyten auftreten, werden durch diese Strukturen gesteuert. Ionenkanäle bilden makromolekulare Komplexe, die aus einer Haupteinheit, die die Kanalpore bildet, und Hilfsproteinen, die diese regulieren, bestehen (Abbildung 1). Die bei LQTS beobachtete Kanalstörung kann an diesen beiden Stellen auftreten: am Hauptprotein oder an den regulierenden Proteinen (Tabelle 1). Die Beteiligung der porenbildenden Einheit, bekannt als alpha, führt zu den drei häufigsten Subtypen von LQTS: LQTS1 (betrifft den Kaliumkanal IKs), LQTS2 (betrifft den Kaliumkanal IKr) und LQTS3 (betrifft den Natriumkanal). Da dies die häufigsten Subtypen sind, sind sie klinisch und genetisch am besten charakterisiert. Die Korrelationen zwischen Phänotyp und Genotyp bei diesen drei Hauptformen sind in Abbildung 2 dargestellt. Das Jervell-Lange-Nielsen-Syndrom entspricht derzeit den Varianten LQTS 1 und 5. Charakteristisch für diese Patienten sind eine angeborene Taubheit und compound homozygote oder heterozygote Mutationen, die den IKs-Strom beeinflussen. Das Romano-Ward-Syndrom umfasst die Varianten von LQTS 1 bis 10 und geht nicht mit Taubheit einher.
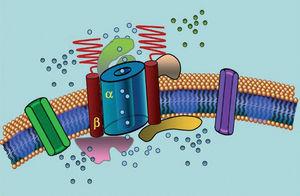
Abbildung 1. Schematische Darstellung des makromolekularen Komplexes. Die Ionenkanäle sind Transmembranproteine (α), die von verschiedenen Proteinen reguliert werden; eines davon ist die sogenannte β-Untereinheit.
Langes QT-Syndrom Typ 1 (LQTS1)
Patienten mit LQTS1 zeigen in der Regel Episoden ventrikulärer Arrhythmien, wenn sie Sport treiben oder einem sympathischen Reiz ausgesetzt sind (68 %).10 Schwimmen wurde als eine Sportart beschrieben, die Arrhythmien bei LQTS1 auslöst.11 Die Penetranz liegt bei diesem Subtyp bei fast 62 %. Die T-Welle hat bei diesen Patienten oft eine breite Basis und eine sehr lange Dauer12,13 (Abbildung 2). Dies ist der häufigste Subtyp und erklärt 30-35 % der Fälle. Das betroffene Gen, KvLQT1 (oder KCNQ1), befindet sich auf Chromosom 11 (11p15.5) und kodiert für die α-Untereinheit des Kaliumkanals IKs. Das Aktionspotenzial wird durch eine Verringerung des ausgehenden K+-Stroms während Phase 3 des Aktionspotenzials verlängert.
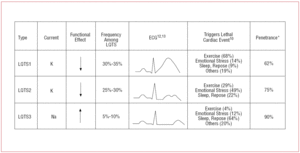
Abbildung 2. Genotyp-Phänotyp-Korrelation bei den häufigsten langen QT-Syndromen. *Bezieht sich auf Fälle, die die Mutation aufweisen und den Phänotyp manifestieren.
Langes QT-Syndrom Typ 2 (LQTS2)
Bei Patienten mit LQTS2 treten ventrikuläre Arrhythmien in der Regel als Reaktion auf emotionalen Stress (49 %) oder plötzliche akustische Reize (z. B. ein Wecker) auf, seltener während des Schlafs (22 %) oder bei körperlicher Betätigung (29 %).10 Frauen in der postpartalen Phase sind besonders anfällig.14 Die geschätzte Penetranz liegt bei 79 %; daher können bis zu 20 % der Fälle ein nicht diagnostisches EKG aufweisen. Die T-Welle bei LQTS2 ist in der Regel niedrigamplitudig und bifid, mit Notching12,13 (Abbildung 2). Das betroffene Gen ist KCNH2 oder HERG auf Chromosom 7 (7q35-36), das für die α-Untereinheit des IKr-Kaliumkanals kodiert und für 25-30 % der Fälle verantwortlich ist. Eine Funktionsstörung dieses Kanals verringert den ausgehenden K+-Strom während der Phase 3 des Aktionspotenzials und verlängert dessen Dauer.
Langes QT-Syndrom Typ 3 (LQTS3)
Patienten mit LQTS3 haben ein höheres Risiko für bösartige Arrhythmien in Ruhe (Schlaf) oder Bradykardie.15 Die Penetranz der SCN5A-Genmutation liegt bei fast 90 %. Das EKG bei LQTS3 zeigt in der Regel eine verzögerte, spitze T-Welle und ermöglicht eine deutliche Beobachtung der ST-Strecken-Verlängerung12,13 (Abbildung 2). Diese Patienten haben in der Regel weniger Symptome als Patienten mit LQTS1 oder LQTS2, aber die Ereignisse sind charakteristischerweise tödlicher.
Das betroffene Gen bei LQTS3 ist SCN5A, das für die α-Untereinheit des Nav1.5-Natriumkanals kodiert (Abbildung 1) und sich auf Chromosom 3 (3p21-24) befindet; es ist in 5-10 % der Fälle die Ursache der Erkrankung. Eine defekte Inaktivierung des Kanals ermöglicht eine anhaltende Zufuhr von Na+ während der Phase 2 des Aktionspotenzials und verlängert dessen Dauer.
Langes QT-Syndrom Typ 4 (LQTS4)
Typ 4 ist eine seltene Variante des LQTS, die fast 1 % der Fälle ausmacht. Es handelt sich um eine atypische Form, die ein breites Spektrum von Arrhythmien hervorruft, darunter katecholaminerge polymorphe ventrikuläre Tachykardie, Vorhofflimmern, intraventrikuläre Überleitungsstörungen, Sinusknoten-Dysfunktion und Bradykardie6-18; außerdem kann der QTc-Wert bei vielen Patienten innerhalb normaler Grenzen liegen. Das betroffene Gen ist ANKB, das sich auf Chromosom 4 (4q25-27) befindet und für die Synthese von Ankyrin-β kodiert, einem Strukturprotein, das die Membranproteine der Kardiomyozyten mit den Proteinen des Zytoskeletts verbindet. Diese Proteine sind die Na/K-ATPase-Pumpe, der Na/Ca-Austauscher und der Inositol-Triphosphat-Rezeptor (InsP3R). Mutationen, die zu einem Verlust der Ankyrin-β-Funktion führen, bewirken einen Anstieg der intrazellulären Kalziumkonzentration und Veränderungen in der Expression der N/K-ATPase und des Na/Ca-Austauschers. Die erhöhte Kalziumkonzentration führt zu frühen und verzögerten Nachdepolarisationen. Somit sind die bei Ankyrin-β-Genmutationen beobachteten ventrikulären Arrhythmien auf spontane Depolarisationen zurückzuführen, die in der Regel als Reaktion auf eine katecholaminerge Stimulation auftreten.
Das Lang-QT-Syndrom Typ 5 (LQTS5)
Typ 5 entsteht durch Veränderungen in der Sequenz des KCNE1-Gens auf Chromosom 21 (21q22.1p22.)19 KCNE1 kodiert für die Synthese der β-Untereinheit des IKs-Kanals, die auch als minK-Untereinheit bezeichnet wird und den IKs-Kanal reguliert. Dieser Typ macht weniger als 1 % der Fälle aus.
Langes QT-Syndrom Typ 6 (LQTS6)
Das betroffene Gen bei Typ 6 ist KCNE2, das sich auf Chromosom 21 (21q22.1) befindet.20 Dieses Gen kodiert für die β-Untereinheit des Kaliumkanals, die auch als MiRP1-Untereinheit bekannt ist, und reguliert den IKr-Kanal. Weniger als 1 % der Fälle sind vom Typ 6.
Langes QT-Syndrom Typ 7 oder Andersen-Tawil-Syndrom (LQTS7)
Die dysmorphen Befunde und elektrokardiographischen Veränderungen, die bei diesem Syndrom auftreten, wurden erstmals 1971 von Dr. Andersen21 beschrieben und 1994 von Dr. Tawil wieder aufgegriffen,22 aber die genetische/molekulare Beschreibung wurde erst 2001 veröffentlicht.23 Das heute als Andersen-Tawil-Syndrom (ATS) bekannte Syndrom ist eine autosomal-dominante Veränderung, die durch periodische Lähmung, abnorme Skelettentwicklung, ventrikuläre Arrhythmie mit häufigen ventrikulären Extrasystolen und eine besondere Anfälligkeit für Kammerflimmern, insbesondere bei Frauen, gekennzeichnet ist. Zu den bei ATS beschriebenen Veränderungen gehören ventrikuläre Extrasystolen (41 %), nicht anhaltende polymorphe ventrikuläre Tachykardien (23 %), bidirektionale ventrikuläre Tachykardien (68 %) und Torsade de pointes (3 %).24 Zu den beobachteten dysmorphen Merkmalen gehören Kleinwuchs, Skoliose, Klinodaktylie, Hypertelorismus, tief angesetzte Ohren, Mikrognathie und eine breite Stirn. Die Ausprägung der Krankheit variiert, was eine frühzeitige Diagnose erschwert.23,25 Mutationen im KCNJ2-Gen auf Chromosom 17 (17q23), das für die Synthese des gleichrichtenden Kaliumkanals Kir 2.1 kodiert, sind für 70 % der Fälle verantwortlich. Dieser Kanal ist an der Phase 4 des Aktionspotenzials beteiligt. Mehrere Autoren stellen die Einbeziehung dieses Gens in die LQTS-Kausalgruppe in Frage, da das QTc-Intervall bei diesem Syndrom nur geringfügig verlängert oder sogar normal ist, die U-Welle jedoch in der Regel ausgeprägt ist, was zu einer Überschätzung des QT-Intervalls geführt hat. Einige Autoren vermuten, dass KCNJ2-Mutationen ATS1 und nicht LQTS7 hervorrufen.24
Das lange QT-Syndrom Typ 8 (LQTS8)
Typ 8 entsteht durch Mutationen im CACNA1-Gen auf Chromosom 12 (12p13.3), das für den L-Typ-Kalziumkanal Cav1 kodiert. 2.Es verursacht das Timothy-Syndrom26 , das durch kardiale Fehlbildungen, intermittierende immunologische Defizite, Hypoglykämie, kognitive Veränderungen einschließlich Autismus, interdigitale Fusion und verlängerte QT-Werte gekennzeichnet ist, die zu Herzrhythmusstörungen und plötzlichem Tod führen.27 Weniger als 0,5 % der Fälle sind vom Typ 8.
Langes QT-Syndrom Typ 9 (LQTS9)
Diese Variante des LQTS entsteht durch Mutationen im CAV3-Gen auf Chromosom 3 (3p25), das für die Caveolin-3-Synthese kodiert. Die Caveola ist eine Einstülpung der Plasmamembran, die an der Endozytose, der Lipidhomöostase und der Signaltransduktion beteiligt ist. Ein wichtiger Bestandteil dieser Struktur ist Caveolin, von dem 3 Subtypen bekannt sind; Subtyp 3 ist spezifisch für Skelett- und Herzmuskel. Einige Ionenkanäle sind in der Caveola angesiedelt, darunter eine kardiale Isoform des Natriumkanals Nav1.5. Vor kurzem wurden mehrere Mutationen in diesem Protein beschrieben. Diese verändern die biophysikalischen Eigenschaften des Natriumkanals Nav1.5 in vitro und führen zu einem Phänotyp, der dem bei LQTS3 beobachteten ähnelt.28 Weniger als 1 % der Fälle werden auf diese Ursache zurückgeführt.
Langes QT-Syndrom Typ 10 (LQTS10)
Typ 10 wurde in einem sehr schweren Fall mit QTc >600 ms, fetaler Bradykardie und 2:1 atrioventrikulärem (AV) Block beschrieben. Sie wird durch Mutationen im SCN4B-Gen verursacht, das sich auf Chromosom 11 (11q23) befindet und für die β4-Untereinheit des Natriumkanals codiert. Es wurden vier verschiedene Subtypen von β-Untereinheiten beschrieben, die interagieren und die verschiedenen Isoformen des Natriumkanals regulieren; dennoch wurde bisher nur der Subtyp 4 mit der Arrhythmogenese in Verbindung gebracht.29 Die Häufigkeit von Mutationen dieses Gens ist nicht untersucht worden, wird aber auf
Mutationen der Jervell-Lange-Nielsen-Variante
Diese schwere Form von LQTS wird durch homozygote30 oder compound heterozygote Mutationen der KCNQ1- und/oder KCNE1-Gene verursacht, die für den IKs-Strom kodieren; es handelt sich also um eine sehr schwere Variante der LQTS1- oder LQTS5-Formen. Diese Erkrankung ist typischerweise mit angeborener Taubheit verbunden. Die Patienten haben in der Regel einen QTc>500 ms und wiederkehrende Synkopen und ein hohes Risiko für einen plötzlichen Tod. Die Eltern von Patienten mit dieser Variante sind in der Regel heterozygot und haben eine weniger schwere Erkrankung oder zeigen keine Symptome.31
DIAGNOSE DES LONG-QT-SYNDROMS
Schwartz-Score
Im Jahr 1985 veröffentlichten Schwartz et al32 die Kriterien für die Diagnose von LQTS, die 1993 modifiziert wurden und wichtige Leitlinien für die Erstbewertung möglicher Fälle enthalten. Dieses System verwendet eine Punktzahl von 1 bis 9 auf der Grundlage der Familienanamnese sowie der klinischen und elektrokardiographischen Befunde. Die Wahrscheinlichkeit einer Erkrankung ist bei einem Score von ≥1 gering, bei 2-3 mittel und bei ≥4 hoch (Tabelle 2).

Pränataldiagnose des Long-QT-Syndroms
Eine fetale Bradykardie kann eine der ersten klinischen Manifestationen des LQTS sein. Retrospektive Serien haben gezeigt, dass bis zu 70 % der Patienten, bei denen im Kindesalter ein LQTS diagnostiziert wird, eine Bradykardie aufweisen, die in der Regel mit einem fetalen Hydrops einhergeht.33 Die Beurteilung der fetalen kardialen Repolarisation zwischen der 14. und 39. Bei dringendem Verdacht auf die Krankheit kann eine Fruchtwasseruntersuchung nach 16 Schwangerschaftswochen für die Diagnosestellung nützlich sein, die leicht zu erreichen ist, wenn ein Elternteil als Träger einer bestimmten Mutation bekannt ist.36
STUDIE EINES PATIENTEN MIT LONG-QT-SYNDROM
Klinische Vorgeschichte
Eine familiäre und/oder persönliche Vorgeschichte mit plötzlichem Herztod ist sowohl für die Diagnose als auch für die Risikostratifizierung von LQTS von entscheidender Bedeutung. Darüber hinaus können auslösende Faktoren und der Kontext der Synkope Hinweise auf den LQTS-Subtyp geben. Bei der Erstuntersuchung eines Verdachtsfalls sollte die Einnahme von Medikamenten, die das QT-Intervall verlängern können, ausgeschlossen werden.
QT-Intervall: Was ist normal?
Das QT-Intervall sollte vorzugsweise in den Ableitungen II oder V5 gemessen werden,37 wo es nachweislich einen größeren prädiktiven Wert hat.38 Dieses Intervall gibt die Dauer der ventrikulären Repolarisation an und wird vom Beginn der Q-Welle bis zum Ende der T-Welle gemessen. Konventionell wird die von Bazett39 vorgeschlagene Formel verwendet, um die Dauer des Intervalls in Abhängigkeit von der Herzfrequenz zu korrigieren (QTc=QT/√RR, ausgedrückt in Sekunden). Obwohl die Messung des QT-Intervalls einfach zu sein scheint, wussten in einer multizentrischen Studie von Viskin et al.40 weniger als 40 % der Ärzte, die keine Kardiologen sind, weniger als 50 % der Kardiologen und mehr als 80 % der Arrhythmie-Spezialisten, wie man es richtig misst. Es ist ratsam, dass Ärzte manuelle Messungen durchführen und sich nicht auf automatische Messungen verlassen, die zwar für andere Intervalle nützlich sein können, aber bei der Berechnung des QT-Intervalls ungenau sind. Das QT-Intervall ist ein dynamisches Intervall, und die normalen Grenzen hängen von mehreren Faktoren ab. Obwohl ein QTc-Intervall von é440 ms bei Männern und é460 ms bei Frauen als abnormal gilt, kann man sowohl Träger von Mutationen als auch gesunde Personen innerhalb dieses Bereichs finden (Abbildung 3). Bei Familien mit LQTS1 wiesen Vincent et al41 nach, dass keiner der Fälle mit positivem Genotyp einen QTc von 470 ms aufwies. Monnig et al38 haben kürzlich gezeigt, dass ein QTc>440 ms ausreicht, um Patienten mit LQTS-assoziierten Mutationen zu erkennen, dass ein QTc>470 ms nützlich ist, um Patienten mit dem Risiko, Symptome zu entwickeln, zu identifizieren, und dass ein QTc>500 ms bei symptomatischen Patienten, die sich einer Behandlung unterziehen, gefunden wird.
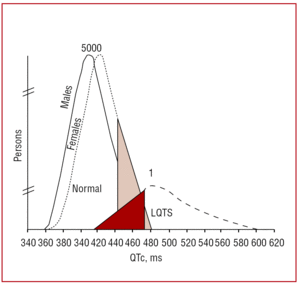
Abbildung 3. Modell der Verteilung des herzfrequenzkorrigierten QT-Intervalls (QTc) bei Patienten mit Mutationen in KVLQT1, HERG oder SCN5A und ihren nicht betroffenen Familienmitgliedern. Die Kurve auf der linken Seite beschreibt die Verteilung der nicht betroffenen Mitglieder und die Kurve auf der rechten Seite die der betroffenen Mitglieder.
Andere elektrokardiografische Veränderungen, die mit dem Long-QT-Syndrom assoziiert sind
Patienten mit LQTS können mehrere T-Wellen-Veränderungen aufweisen: Polaritätsalternans, Amplitudenvariationen, Notching und ein biphasisches Erscheinungsbild, unter anderem.42 T-Wellen-Alternans (Abbildung 4A) ist definiert als eine schlagweise Variation der Amplitude, Morphologie und Polarität einer Sinusrhythmus-T-Welle, ohne Variationen im QRS-Komplex. Sie ist ein Indikator für elektrische Instabilität,43 die eine regionale Dispersion der ventrikulären Repolarisation widerspiegelt und gelegentlich einem Kammerflimmern vorausgeht.44
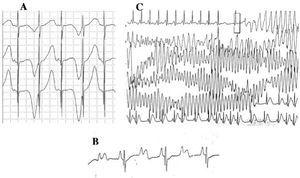
Abbildung 4. Elektrokardiographische Veränderungen beim langen QT-Syndrom. A: Elektrische T-Wellen-Alternans. B: Atrioventrikulärer Block 2:1. C: Selbstbegrenzte Torsade de pointes.
Patienten mit LQTS können Anzeichen einer Sinusknoten-Dysfunktion, Bradykardie und/oder Pausen aufweisen.45 Die Subtypen LQTS1 und LQTS3, insbesondere der letztere, weisen häufig eine Sinusbradykardie auf,46 während LQTS4 mit einer Sinusknotendysfunktion in Verbindung gebracht wurde.18
Seit dem Jahrzehnt 1970-1980 wurde die Koexistenz von AV-Leitungsstörungen mit LQTS47 beobachtet (Abbildung 4B). Der Zwei-zu-Eins-AV-Block ist eine seltene Manifestation mit schlechter Prognose, die bereits im fetalen Stadium in Form einer persistierenden Bradykardie auftreten kann. Die Inzidenz dieser Anomalie wurde mit 4-5 % angegeben48 und ist trotz der Behandlung mit Betablockern und/oder Herzschrittmachern mit einer hohen Sterblichkeit verbunden.49,50 Dieses Phänomen lässt sich durch eine lange Dauer des Aktionspotenzials erklären. Wenn die ventrikuläre Refraktärzeit verlängert ist, wird der nachfolgende Impuls der Sinusaktivität blockiert, weil er die Ventrikel erreicht, wenn sie sich noch in der Refraktärzeit befinden. Diese Veränderung scheint ausschließlich bei LQTS aufzutreten, da die ventrikuläre Refraktärperiode länger ist als die des AV-Leitungssystems.51 Die Steigung des QRS-Komplexes ist in der Regel steil und der Block wurde im Infra-His-Bereich lokalisiert,46,51,52 aber der Ort des Blocks kann vom Genotyp abhängen. Bisher wurden 4 Gene mit dem 2:1-Block bei LQTS in Verbindung gebracht: HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 und SCN4B (LQTS10).55
Die charakteristische ventrikuläre Arrhythmie von LQTS ist als Torsade de pointes bekannt (Abbildung 4C). Sie tritt auf, wenn das QT-Intervall unabhängig von der Ätiologie verlängert ist. Es handelt sich um eine polymorphe ventrikuläre Tachykardie aufgrund von Reentry, die elektrokardiografisch durch eine kontinuierliche Drehung der QRS-Achse um eine imaginäre Linie gekennzeichnet ist. In der Regel geht ihr eine Pause voraus, gefolgt von einer Extrasystole (kurz-lang-kurzes RR-Intervall), wie in der Abbildung dargestellt.56-58 Sie kann in Kammerflimmern und plötzlichem Tod enden. Wenn dies nicht der Fall ist, kann der Patient nur eine Synkope erleiden, und wenn die Episode kurz ist, kann sie unentdeckt bleiben.
Holter
Holter-Studie bietet eine vollständige, dynamische Bewertung des QT-Intervalls. Gelegentlich werden spontane Episoden asymptomatischer ventrikulärer Arrhythmien aufgezeichnet, ebenso wie Episoden einer Sinusknoten-Dysfunktion oder eines AV-Blocks.
Belastungsuntersuchung
Patienten mit LQTS können die altersabhängig berechnete maximale Herzfrequenz nicht erreichen. Darüber hinaus kann das QT-Intervall unter Belastung ein paradoxes Verhalten zeigen, indem es sich eher vergrößert als verkleinert.59,60 Das elektrokardiografische Muster während des Belastungstests ist je nach Art des LQTS unterschiedlich. Patienten mit LQTS1 erreichen nicht nur nicht die für ihr Alter berechnete maximale Herzfrequenz, sondern weisen auch häufig ein verlängertes QT-Intervall auf, während Patienten mit LQTS2 ihre erwartete Herzfrequenz erreichen können und nur einen leichten oder gar keinen Anstieg des QT-Intervalls zeigen.61,62 Im Allgemeinen zeigen Patienten mit LQTS3 eine physiologische Reaktion auf Belastung, d. h. eine normale Verkürzung des QT-Intervalls.63 Stresstests können auch nützlich sein, um das Ansprechen auf die Behandlung zu beurteilen und das Risiko in asymptomatischen Fällen oder bei Zweifeln an den Ereignissen, die zur Arrhythmie führen, zu stratifizieren.
Genetisches Screening
In den letzten Jahren waren genetische Studien bei LQTS auf Forschungslabors beschränkt. Dennoch waren die daraus gewonnenen Informationen für die Behandlung von Patienten, insbesondere von Hochrisikopatienten, äußerst nützlich. Die vielleicht wichtigste Anwendung des Screenings ist die genetische Beratung, aber es hat auch wichtige Auswirkungen auf die Behandlung, die sich nach dem betroffenen Kanal richten kann. Die genaue Lokalisierung einer bestimmten Mutation kann zusätzliche Informationen über die Entwicklung des Risikos liefern. Patienten mit Mutationen in der Transmembranregion von KCNQ1 (IKS) haben eine größere Wahrscheinlichkeit für das Auftreten von Arrhythmien als solche mit Mutationen in der C-terminalen Region64; dasselbe gilt für Patienten mit Mutationen in der Porenregion von KCNH2 oder HERG65 im Vergleich zu solchen mit Mutationen in der N- oder C-terminalen Region.66
Das anfängliche Screening kann vielleicht auf die Gene KCNQ1, HERG und SCN5A beschränkt werden, die in 65 % der Fälle die Möglichkeit bieten, Mutationen zu finden. Bei negativen Ergebnissen kann das Screening auf die Gene KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 und SCN4B ausgedehnt werden, was die Wahrscheinlichkeit positiver Ergebnisse um 5 % bis 10 % erhöht.
Postmortales genetisches Screening
Interessant ist, dass Genmutationen, die zu LQTS führen, bei Kindern, die einen plötzlichen Tod erlitten haben, und bei unerklärlichen Fällen von plötzlichem Tod bei jungen Erwachsenen gefunden wurden.
In postmortalen genetischen Studien an Patienten mit plötzlichem Tod und negativer Autopsie wurden Mutationen, die zu LQTS führen, in unterschiedlichen Prozentsätzen nachgewiesen67-69: fast 10 % bei Kindern und 35 % bei jungen Erwachsenen.70-72 Aufgrund dieser Ergebnisse wurde eine routinemäßige EKG-Untersuchung bei allen Neugeborenen vorgeschlagen.73,74
Die postmortale genetische Untersuchung, die in der Literatur auch als „molekulare Autopsie“ bezeichnet wird, hat nicht nur rechtliche Konsequenzen, sondern auch wichtige Auswirkungen auf Familien, die möglicherweise betroffen sind, ohne es zu wissen.
Regulatorische Polymorphismen
In der LQTS-Population wurden mehrere häufig vorkommende Polymorphismen beschrieben, die sich auf fast alle mit dieser Erkrankung assoziierten Gene verteilen. Obwohl diese Veränderungen offenbar nicht pathogen sind, können einige die folgenden Auswirkungen haben75-78:
1. Erzeugung einer individuellen Anfälligkeit für die Entwicklung von Herzrhythmusstörungen.
2. Begünstigung der pathogenen Wirkung einer anderen nicht-synonymen Veränderung.
3. Verringerung der pathogenen Wirkung einer anderen nicht-synonymen Veränderung.
Dies ist der Fall beim K897T-Polymorphismus in KCNH2 (HERG), der bei bis zu 15 % der Bevölkerung vorkommt und nicht nur mit der Anfälligkeit für bestimmte Medikamente in Verbindung gebracht wird,79 sondern auch die pathogene Wirkung von Mutationen in demselben Gen begünstigt.78 Ein weiteres Beispiel ist der S1103Y-Polymorphismus im SCN5A-Gen, der vor allem bei Schwarzen vorkommt, eine Inzidenz von fast 13 % aufweist und mit einem erhöhten Risiko eines plötzlichen Todes im Kindesalter verbunden ist.80
Interessanterweise wurden im Produkt des SCN5A-Gens (das für die Nav1.5-Natriumkanal-Isoform beim Menschen kodiert) zwei alternative Prozessierungsstellen beschrieben, die zwei Arten von Natriumkanälen erzeugen: eine mit 2016 Aminosäuren, die Glutamin in der Position 1077 (Q1077) enthalten, und eine andere mit 2015 Aminosäuren, denen Glutamin fehlt (Q1077del). Transkripte dieser alternativen Verarbeitungen sind in einem Verhältnis von 2:1 im selben menschlichen Herzen vorhanden, und mehrere häufige Polymorphismen haben unterschiedliche Auswirkungen auf die Kanalfunktion, je nachdem, ob der Kontext Q1077 oder Q1077del ist. Dies wurde zunächst anhand des H558R-Polymorphismus von SCN5A gezeigt, der bei bis zu 30 % der Bevölkerung vorkommt. Wurde H558R im Kontext von Q1077 exprimiert, wurde eine starke Verringerung des Ionenstroms beobachtet.81 Ein ähnlicher Effekt wurde für den Polymorphismus S524Y82 dokumentiert. Diese Befunde haben Faktoren geliefert, die den unterschiedlichen Schweregrad der Krankheit sowie die in einigen Familien beobachteten unterschiedlichen Phänotypen derselben Mutation erklären.77
Pharmakologische Tests mit Adrenalin
Pharmakologische Tests mit niedrig dosiertem Adrenalin sind eine sichere und nützliche Option, um Verdachtsfälle von LQTS mit einem grenzwertigen QTc zu entlarven. Mit einer Sensitivität von 92,5 %, einer Spezifität von 86 %, einem positiven prädiktiven Wert von 76 % und einem negativen prädiktiven Wert von 96 % ist dieser Test besonders wirksam bei der Erkennung asymptomatischer Formen von LQTS1. Er kann auch für die Diagnose von LQTS2 nützlich sein, allerdings mit geringerer Sensitivität und Spezifität. Bei LQTS3 oder anderen Formen von LQTS ist er nicht hilfreich. Unter normalen Bedingungen induziert die Stimulation des Sympathikus eine Phosphorylierung des IKs-Kaliumkanals, wodurch seine Funktion optimiert wird und eine Verkürzung des Aktionspotenzials eintritt. Bei Patienten mit LQTS, insbesondere Typ 1, wird eine paradoxe Reaktion auf die Verabreichung von niedrig dosiertem Adrenalin (0,025-0,2 µg/kg/min) beobachtet, die das QT-Intervall auf mehr als 30 ms83-86 verlängert.
QT-INTERVALL-VERLÄNGERUNG UND DURCH ARZNEIMITTEL VERURSACHTE TORSADE DE POINTES
Eine Vielzahl von Arzneimitteln, die in verschiedenen medizinischen Fachbereichen verwendet werden, kann eine iatrogene Verlängerung des QT-Intervalls verursachen. Einige Medikamente wurden wegen dieser unerwünschten Wirkung vom Markt genommen (z. B. Astemizol und Cisaprid; weitere Informationen unter www.qtdrugs.org).87,88
Ventrikuläre Arrhythmien als Folge von nicht-antiarrhythmischen Medikamenten treten bei weniger als einem von 10 000 bis 100 000 exponierten Personen auf. Wenn man bedenkt, dass klinische Studien zwischen 2000 und 3000 Probanden umfassen, würde diese unerwünschte und tödliche Nebenwirkung in der klinischen Phase der Arzneimittelentwicklung leicht unentdeckt bleiben.89 Dieser Punkt hat ein enormes Interesse an Sicherheitsaspekten bei der Erforschung und Entwicklung neuer Medikamente geweckt.
Zu den Faktoren, die mit der individuellen Anfälligkeit zusammenhängen, gehören das weibliche Geschlecht, Hypokalzämie, Hypomagnesiämie, Bradykardie, Herzinsuffizienz, Postkardioversion, Vorhofflimmern, linksventrikuläre Hypertrophie, unerkanntes LQTS, prädisponierende Polymorphismen und hohe Serumkonzentrationen prädisponierender Medikamente.90
Der Kanal, der typischerweise mit Medikamenten interagiert, ist IKr, der durch das KCNH2(HERG)-Gen kodiert wird, aufgrund seiner Molekularstruktur. Andere Kaliumkanäle haben 2 Prolinreste, die zur Kanalpore hin abgewinkelt sind und das Lumen verkleinern. Im Gegensatz dazu fehlen bei IKr diese Reste, wodurch ein größerer Porenvorraum entsteht und der Kontakt mit großen Molekülen erleichtert wird. Darüber hinaus verfügt er über zwei aromatische Reste (Tyrosin und Phenylalanin), die die Bindung mit aromatischen Molekülen begünstigen, die in mehreren Medikamenten enthalten sind, die den Kanal blockieren können.91
Wie bereits erwähnt, ist die LQTS-Penetranz unvollständig, und einige asymptomatische Träger von Mutationen können nach Verabreichung eines dieser Medikamente bösartige Herzrhythmusstörungen entwickeln. Darüber hinaus verleihen Polymorphismen, die in der Bevölkerung als häufig gelten, eine individuelle Anfälligkeit für die Entwicklung von Torsade de pointes, wenn bestimmte Medikamente eingenommen werden. Dies ist der Fall beim Polymorphismus R1047L, dem zweithäufigsten Polymorphismus in KCNH2, der mit der Entwicklung von Torsade de pointes bei der Einnahme des Medikaments Dofetilid in Verbindung gebracht wurde.92 Mindestens 20 Polymorphismen des Gens KCNH2 wurden bei gesunden Personen beschrieben, und ihre Auswirkungen auf die individuelle Anfälligkeit für die Entwicklung arzneimittelbedingter Herzrhythmusstörungen müssen noch ermittelt werden.93 Polymorphismen, die eine Anfälligkeit für die Entwicklung ventrikulärer Herzrhythmusstörungen vermitteln, wurden auch beim Natriumkanal Na1.5 nachgewiesen. Dies gilt für den Polymorphismus H558R, der bei bis zu 30 % der Bevölkerung vorkommt, oder S1103Y, der bei Schwarzen häufig ist80,81,90,94,95; Ihre Bedeutung für die arzneimittelinduzierte Anfälligkeit ist nicht untersucht worden.
LONG QT SYNDROM UND SCHWANGERSCHAFT
Genetische Beratung ist bei LQTS wichtig, aber im Allgemeinen gibt es keine Kontraindikation für eine Schwangerschaft bei Frauen, die Trägerinnen sind, obwohl jeder Fall anders ist und individuell in dem entsprechenden Kontext bewertet werden sollte.
Es wurde festgestellt, dass das Risiko einer bösartigen ventrikulären Arrhythmie mit der Schwangerschaft abnimmt. Im Gegensatz dazu wurde innerhalb der ersten 9 Monate nach der Entbindung eine größere Anfälligkeit für bösartige Herzrhythmusstörungen festgestellt, insbesondere bei Patienten mit LQTS2. Dieses Risiko nimmt mit einer Betablockertherapie erheblich ab.96
RISIKOSTRATIFIKATION
Die Entwicklung von LQTS ist unterschiedlich und wird von der Dauer des QTc-Intervalls, Umweltfaktoren, Alter, Genotyp und Ansprechen auf die Behandlung beeinflusst.97,98 Ventrikuläre Arrhythmien treten häufiger bei LQTS1 und LQTS2 auf, sind aber bei LQTS3 schwerer.99 Wie bereits erwähnt, sind Frauen in der Zeit nach der Geburt besonders anfällig für bösartige Arrhythmien.14
Das lange QT-Syndrom sollte als Hochrisikofaktor betrachtet werden, wenn es mit Folgendem verbunden ist:
1. Angeborene Taubheit (Jervell-Lange-Nielsen-Syndrom).
2. Wiederkehrende Synkope aufgrund einer malignen ventrikulären Tachyarrhythmie.
3. plötzlicher Tod in der Familie.
4. QTc>500 ms.
5. 2:1 atrioventrikulärer Block.
6. Elektrische T-Wellen-Alternans.
7. LQTS3-Genotyp.
Die Studie von Priori et al97, die an 647 Patienten durchgeführt wurde, hat gezeigt, dass die Wahrscheinlichkeit, ein schwerwiegendes Ereignis (Synkope, Herzstillstand, plötzlicher Tod) vor dem 40. Lebensjahr zu erleiden, hoch ist (>50 %), wenn der QTc >500 ms bei LQTS1, LQTS2 und bei Männern mit LQTS3 beträgt. Kürzlich wurde über eine Analyse des internationalen LQTS-Registers berichtet. Das Risiko eines plötzlichen Todes wurde bei 2772 Jugendlichen mit dieser Krankheit analysiert, und es wurden 3 Faktoren ermittelt, die mit einem höheren Risiko in dieser Population verbunden sind: QTc>530 ms, Synkopen in den letzten 10 Jahren und Geschlecht; 10- bis 12-jährige Jungen hatten ein höheres Risiko als Mädchen, aber in der Altersgruppe von 13 bis 20 Jahren war das Risiko vergleichbar.100
Behandlung
Symptomatische Patienten, die keine Behandlung erhalten, haben eine jährliche Sterblichkeitsrate von 20 % und eine 10-Jahres-Sterblichkeit von 50 % nach dem ersten Auftreten einer ventrikulären Arrhythmie. Obwohl klar ist, dass bei Vorliegen von Symptomen eine Behandlung eingeleitet werden sollte, ist die Vorgehensweise bei asymptomatischen Patienten noch umstritten. Es wurde dokumentiert, dass bei 9 % der Patienten ein Herzstillstand die erste Manifestation der Erkrankung sein kann,48 und dass 12 % der asymptomatischen Patienten Symptome entwickeln und möglicherweise einen plötzlichen Tod erleiden. Bei allen Patienten mit LQTS sollte eine Erstbehandlung mit Betablockern eingeleitet werden. Eine Einschränkung der körperlichen Belastung ist empfehlenswert, aber die klinischen und elektrokardiografischen Risikomarker sind eine nützliche Entscheidungsgrundlage. Es ist wichtig, die Patienten über das Risiko der Einnahme verschiedener Medikamente zu informieren, die das QT-Intervall verlängern und die Entwicklung ventrikulärer Arrhythmien begünstigen können, wie bereits erwähnt. Die genetische Diagnose ermöglicht nicht nur eine angemessene Beratung der Familie im Zusammenhang mit der Krankheit, sondern hilft auch bei der Einschätzung der Prognose und der Ausrichtung der spezifischen Behandlung.
Betablocker
Betablocker sind die Behandlung der ersten Wahl bei LQTS und sollten bei allen Patienten als Erstbehandlung eingesetzt werden.101 Sie senken das Risiko kardiovaskulärer Ereignisse um bis zu 64 %100 und sind besonders wirksam bei Patienten mit IKs-Kanal-Mutationen (LQTS1),102 die zu einem großen Teil durch das sympathische System reguliert werden. Betablocker verändern nicht das QT-Intervall, sondern dessen Streuung.103 Obwohl diese Medikamente die Häufigkeit von Ereignissen verringern,104,105 hat sich gezeigt, dass 10 % der Patienten mit LQTS1, 23 % mit LQTS2 und 32 % mit LQTS3 trotz Behandlung kardiovaskuläre Symptome haben.106 Insbesondere Patienten mit LQTS3 scheinen keine nennenswerten Vorteile zu erzielen; tatsächlich sollte diese Medikamentengruppe bei diesen Patienten mit Vorsicht eingesetzt werden, da Episoden von ventrikulären Arrhythmien bei LQTS3 häufiger auftreten, wenn die Herzfrequenz niedrig ist. Im Allgemeinen werden 32 % der symptomatischen Patienten in den ersten 5 Jahren vor Beginn einer Betablocker-Behandlung wiederkehrende Symptome haben, und 14 % der Patienten, die nach einem plötzlichen Herztod gerettet wurden, werden innerhalb von 5 Jahren ein weiteres ähnliches Ereignis erleiden, wenn sie nur diese Therapie erhalten.107 Bei der Behandlung von LQTS werden verschiedene Betablocker eingesetzt, hauptsächlich Nadolol (0,5-1 mg/kg/Tag), Propranolol (2-4 mg/kg/Tag), Metoprolol (0,5-1 mg/kg/Tag) und Atenolol (0,5-1 mg/kg/Tag). Atenolol ist bei LQTS jedoch möglicherweise nicht vorteilhaft; es wurde mitgeteilt, dass mindestens 75 % der Patienten, die nicht auf eine Betablocker-Therapie ansprachen, Atenolol erhielten, obwohl dieses Ergebnis möglicherweise mit der Verwendung suboptimaler Dosen zusammenhängt.104 Belastungstests sind nützlich, um die geeignete Dosis zu ermitteln. Die maximale Herzfrequenz sollte während der Behandlung 130 Schläge/min nicht überschreiten.
Natriumkanalblocker
Natriumkanalmutationen, die LQTS3 verursachen, führen zu einer defekten Inaktivierung des Kanals; eine Natriumkanalblockade hat sich bei diesen Patienten als nützlich erwiesen. Studien, die mit Flecainid durchgeführt wurden, haben Verbesserungen der Herzfrequenz, der T-Wellen-Veränderungen und des QT-Intervalls dokumentiert.108 Auch von Mexiletin wurde berichtet, dass es die elektrokardiografischen Risikomarker verbessert.63,109,110 In-vitro-Studien mit Ranolazin haben gezeigt, dass die schädlichen Auswirkungen der Mutationen, die beim Menschen festgestellt wurden, abnehmen.111 Obwohl die Ergebnisse ermutigend sind, sollte bedacht werden, dass es keine Langzeitstudien gibt, die diese Therapie bewerten, und dass keine Ergebnisse aus großen Serien vorliegen. Natriumkanalblocker sollten nicht verabreicht werden, wenn keine gesicherte genetische Diagnose vorliegt.
Kaliumergänzung und Medikamente, die seine Verfügbarkeit erhöhen
Kaliumergänzungen und/oder kaliumsparende Medikamente, wie Spironolacton, verkürzen das QTc-Intervall in 24 % der Fälle.112,113 Medikamente, die die Öffnung der Kaliumkanäle begünstigen, wie Aprikalim, Levcromakalim, Nicorandil und Pinacidil, haben sich als nützlich für die Behandlung von LQTS erwiesen. Die Subtypen, bei denen sie von besonderem Nutzen sind, sind LQTS1 und LQTS2.114
Herzschrittmacher und Defibrillatoren
Die Stimulation von Herzschrittmachern wurde bei Patienten mit pausenabhängiger Arrhythmie eingesetzt.115,116 Patienten mit LQTS3 profitieren in der Regel stärker von dieser Behandlung, da die Prävalenz von Bradykardie in dieser Gruppe größer ist. Die DDD-Stimulation ist bei Patienten mit pausenabhängiger Arrhythmie oder hochgradigem 2:1-AV-Block indiziert. Frequenzen, die auf weniger als 70 Schläge/Minute117 programmiert sind, sind nicht geeignet, um ventrikuläre Arrhythmien zu verhindern. Es wird empfohlen, den Sensor auf schnelles Ansprechen zu programmieren, da diese Patienten in der Regel eine unangemessene Beschleunigung der Herzfrequenz als Reaktion auf eine Belastung aufweisen. Alle Funktionen, die das Vorhandensein von Pausen implizieren, sollten ausgeschaltet werden, wie z. B. die Hysterese- und die nächtliche Funktion. Die PARP (postventrikuläre atriale Refraktärzeit) sollte so kurz wie möglich sein. Die Frequenzregulierungsfunktion sollte eingeschaltet sein, um eine postextrasystolische Pause zu verhindern. Es ist zu bedenken, dass auch ein Oversensing der T-Wellen und Capture-Fehler zu Pausen führen können. Der kombinierte Einsatz eines implantierbaren Kardioverter-Defibrillators (ICD) und von Betablockern senkt die Inzidenz des plötzlichen Herztodes erheblich.118-120 Die Indikation für diese Maßnahmen ist in Hochrisikofällen eindeutig gegeben.121 Die Programmierung des Geräts hängt von den Bedürfnissen des einzelnen Patienten ab, aber im Allgemeinen sollte die Verabreichung einer Behandlung bei asymptomatischen, selbstbegrenzten Ereignissen vermieden werden; zu diesem Zweck ist eine Erkennungszeit von 15 s angezeigt. Arrhythmische Stürme sind eine Komplikation der AID-Therapie. Bei fast 15 % der Patienten kann es zu dieser Komplikation kommen, die zum großen Teil auf einen erhöhten Sympathikustonus nach dem ICD-Schock zurückzuführen ist.118 Dieses Problem kann durch eine Erhöhung der Betablocker-Dosis in den Griff bekommen werden. Wenn diese Maßnahme nicht hilfreich ist, sollte eine Resektion der sympathischen Kettenganglien in Betracht gezogen werden.
Linke Sympathektomie
1971 wurde die sympathische Gangliektomie als nützliche therapeutische Option bei diesen Patienten eingeführt.122 1991 veröffentlichten Schwartz et al123 die erste Serie von 85 Patienten mit schlechtem Ansprechen auf die Betablocker-Behandlung, bei denen eine linke Gangliektomie mit ermutigenden Ergebnissen durchgeführt wurde: eine 5-Jahres-Überlebensrate von 94 %. Derzeit wird diese therapeutische Option Hochrisikopatienten angeboten, die trotz Betablocker-Behandlung und/oder Schrittmacher-Implantation weiterhin an Synkopen leiden, sowie Patienten, die häufig Schocks von ihrem implantierten Defibrillator erhalten. Das Verfahren besteht in der Resektion des unteren Teils des Ganglion stellatum und der linken Thoraxganglien T2 bis T4 der Sympathikuskette, da sich eine einfache linke Stilektomie als nicht ausreichend wirksam erwiesen hat. Die mikroinvasive Thorakoskopie124,125 ist mit guten Ergebnissen eingesetzt worden. Die größte Serie von Patienten, die mit dieser Methode behandelt wurden, wurde kürzlich veröffentlicht und zeigte eine signifikante Verringerung der Anzahl von Synkopen oder plötzlichen Todesfällen sowie eine 5-Jahres-Überlebensrate von 95 %. Bei Patienten mit früheren Synkopen lag die 5-Jahres-Überlebensrate bei 97 %, wobei die Wahrscheinlichkeit eines erneuten Auftretens bei 11 % lag, wobei es sich in der Mehrzahl der Fälle um ein einziges synkopisches Ereignis handelte. Nach der linken Sympathektomie kam es außerdem zu einer signifikanten Verkürzung des QT-Segments. Trotz dieser günstigen Ergebnisse ist die Prävention des plötzlichen Todes nicht vollständig, sondern konnte auf 3 % reduziert werden. Bei Patienten mit einem ICD, die wegen multipler Defibrillatorschocks operiert wurden, sank die durchschnittliche Anzahl der Ereignisse von 25 auf 0, was einer Verringerung um 95 % entspricht. Ein positiver Effekt wurde bei LQTS1 bestätigt. Bei Patienten mit LQTS2 ist der Nutzen wahrscheinlich geringer, und bei LQTS3 ist die Wirksamkeit nicht erwiesen.126
Ablation
Es wurde berichtet, dass die Ablation der Extrasystole, die in einigen Fällen die ventrikuläre Arrhythmie auslöst, mit einer Verringerung der Häufigkeit von Episoden einhergehen kann.127 Es gibt jedoch keine Langzeitstudien mit einer angemessenen Anzahl von Patienten, um den routinemäßigen Einsatz dieser Technik zu rechtfertigen.
Siehe Leitartikel auf den Seiten 675-82
ABKÜRZUNGEN
AV: atrioventrikulär
AID: automatischer implantierbarer Defibrillator
ECG: Elektrokardiogramm
QTc: herzfrequenzkorrigiertes QT
ATS: Andersen-Tawil-Syndrom
LQTS: langes QT-Syndrom
Dr. Medeiros erhält wirtschaftliche Unterstützung von CONACyT und FUNSALUD.