INTRODUCTION
Le syndrome du QT long (LQTS) se caractérise par une repolarisation ventriculaire gravement altérée, entraînant un allongement de l’intervalle QT à l’électrocardiogramme (ECG). Cette affection prédispose les patients à l’arythmie ventriculaire maligne (torsade de pointes) et à la mort subite. La description clinique et électrocardiographique du syndrome du QT long a été rapportée en 1957 par Anton Jervell et Fred Lange Nielsen1, qui ont publié leurs études sur une famille de parents non-consanguins comptant 6 enfants. Quatre des enfants présentaient une surdité congénitale et des épisodes syncopaux, et 3 présentaient une mort subite. L’étude ECG de ces patients a montré un intervalle QT anormalement long. Les deux parents étaient asymptomatiques, avaient un ECG normal et ne présentaient aucun problème d’audition. En 1964, Romano et Ward ont signalé indépendamment un syndrome cardiaque caractérisé par des syncopes récurrentes, des antécédents familiaux de mort subite et un allongement de l’intervalle QT sans surdité neuronale.2 Des études génétiques ultérieures ont montré que le syndrome décrit par Jervell et Lange Nielsen, qui s’accompagne d’une surdité neuronale congénitale, correspond à des mutations homozygotes, avec un phénotype sévère et un risque élevé de mort subite. L’affection connue sous le nom de syndrome de Romano-Ward correspond généralement à des mutations hétérozygotes, les patients ne présentent pas d’altérations auditives et la gravité de la maladie varie considérablement. Près d’un demi-siècle plus tard, en 19953,4, les principaux gènes associés au LQTS ont été décrits et la maladie a été reconnue comme un trouble des canaux ioniques cardiaques. Il s’agit de la première canalopathie cardiaque à être décrite et c’est peut-être le trouble des canaux ioniques arythmogènes le plus étudié à ce jour. Le tableau clinique est très variable : le patient peut être asymptomatique ou présenter des syncopes récurrentes, des crises d’épilepsie ou une mort subite comme première manifestation de la maladie. Au départ, le LQTS était considéré comme un syndrome rare et, de fait, la présentation sévère de la maladie est sporadique. Néanmoins, l’incidence des mutations apparentées est estimée à 1/3000-5000 cas,5 32% des porteurs asymptomatiques peuvent avoir un intervalle QT corrigé de la fréquence cardiaque (QTc) dans les limites normales, la maladie est transmise à 50% de leurs descendants, ils sont plus susceptibles de développer une arythmie par rapport à la population générale, et jusqu’à 20% peuvent devenir symptomatiques.6
Le syndrome du QT long présente une grande hétérogénéité génétique. Plus de 500 mutations réparties dans 10 gènes ont été décrites dans cette affection : KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 et SCN4B. Malgré les progrès réalisés dans ce domaine, un diagnostic génétique ne peut être établi chez 25 à 30 % des patients.7,8 La présentation de la maladie est principalement monogénique6 ; les variétés polygéniques ou composites présentent généralement un phénotype plus sévère. La pénétrance, c’est-à-dire le nombre de patients qui présentent la mutation et manifestent le phénotype, varie de 25 à 90 %.9 Moins fréquemment, il peut y avoir des variations dans l’expressivité de la maladie, plusieurs phénotypes résultant de la même mutation. Les études de génétique moléculaire développées au cours des 11 dernières années ont permis d’établir d’importantes corrélations génotype-phénotype, qui ont contribué à orienter l’approche thérapeutique. En outre, des observations intéressantes ont été faites sur la susceptibilité individuelle à développer une arythmie dans les études portant sur les polymorphismes non synonymes fréquents dans cette population, un aspect qui a suscité un intérêt considérable, notamment dans le domaine de la pharmacogénomique.
CLASSIFICATION DU SYNDROME DU LONG QT
Concepts généraux
La classification du LQTS utilisée dans le passé était basée sur la présentation homozygote ou hétérozygote de la maladie, qui donne lieu respectivement au syndrome de Jervell-Lange-Nielsen (avec surdité) et au syndrome de Romano-Ward (sans surdité). La présente classification met l’accent sur les résultats génétiques, comme l’illustre le tableau 1. Les 3 principaux gènes associés à la maladie ont été décrits en 1995-1996. Ces gènes, qui codent pour les unités de formation des pores des canaux potassiques IKs et IKr, et du canal sodique Nav1.5, représentent près de 65 % des cas. Bien que, les années suivantes, sept autres gènes aient été inclus dans la liste, ils ne représentent que 5 % des cas.
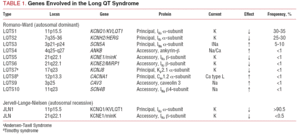
Les canaux ioniques sont des protéines transmembranaires qui transportent les ions à travers la membrane cellulaire. Les canaux impliqués dans le LQTS sont sélectifs ou spécialisés dans le transport d’un seul ion et sont voltage-dépendants, c’est-à-dire que leur activation se produit à une tension intracellulaire spécifique, qui varie selon le sous-type de canal. Les phénomènes électriques et contractiles qui se produisent dans le cardiomyocyte sont contrôlés par ces structures. Les canaux ioniques forment des complexes macromoléculaires constitués d’une unité principale qui forme le pore du canal et de protéines auxiliaires qui le régulent (Figure 1). Le dysfonctionnement des canaux observé dans le LQTS peut se produire à ces deux endroits : la protéine principale ou les protéines régulatrices (tableau 1). L’implication de l’unité de formation du pore, appelée alpha, génère les trois sous-types les plus courants de LQTS : LQTS1 (affectant le canal potassique IKs), LQTS2 (affectant le canal potassique IKr) et LQTS3 (affectant le canal sodique). Comme il s’agit des sous-types les plus fréquents, ils sont les mieux caractérisés cliniquement et génétiquement. Les corrélations phénotype-génotype dans ces trois formes principales sont décrites dans la figure 2. Actuellement, le syndrome de Jervell-Lange-Nielsen correspond aux variétés LQTS 1 et 5. De manière caractéristique, ces patients présentent une surdité congénitale et des mutations homozygotes ou hétérozygotes composées qui affectent le courant IKs. Le syndrome de Romano-Ward comprend les variétés de LQTS 1 à 10 et ne comporte pas de surdité.
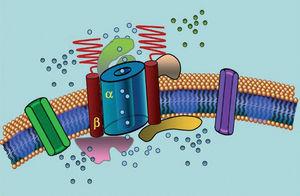
Figure 1. Représentation schématique du complexe macromoléculaire. Les canaux ioniques sont des protéines transmembranaires (α) régulées par diverses protéines ; l’une d’entre elles est la sous-unité dite β.
Syndrome du QT long de type 1 (LQTS1)
Les patients atteints du LQTS1 présentent généralement des épisodes d’arythmie ventriculaire lors d’un exercice physique ou lors d’un stimulus sympathique (68%).10 La natation a été décrite comme un sport déclenchant l’arythmie dans le LQTS1.11 La pénétrance est de près de 62% dans ce sous-type. L’onde T chez ces patients a souvent une base large et une durée très prolongée12,13 (Figure 2). Il s’agit du sous-type le plus fréquent et explique 30 à 35 % des cas. Le gène affecté, KvLQT1 (ou KCNQ1), est situé sur le chromosome 11 (11p15.5) et code pour la sous-unité α du canal potassique IKs. Le potentiel d’action est prolongé par une réduction du courant K+ sortant pendant la phase 3 du potentiel d’action.
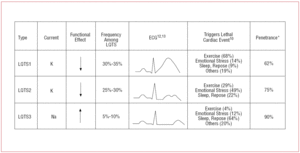
Figure 2. Corrélation génotype-phénotype dans les syndromes du QT long les plus fréquents. *Les patients atteints du syndrome du QT long de type 2 (LQTS2) ont tendance à présenter une arythmie ventriculaire en réponse à un stress émotionnel (49 %) ou à des stimuli auditifs soudains (par exemple, un réveil), et moins fréquemment pendant le sommeil (22 %) ou l’exercice physique (29 %).10 Les femmes en période post-partum sont particulièrement sensibles à ce type de syndrome. La pénétrance estimée est de 79 % ; par conséquent, jusqu’à 20 % des cas peuvent présenter un ECG non diagnostique. L’onde T du LQTS2 est généralement de faible amplitude et bifide, avec une encoche12,13 (figure 2). Le gène affecté est KCNH2 ou HERG, situé sur le chromosome 7 (7q35-36), qui code pour la sous-unité α du canal potassique IKr et représente 25 à 30 % des cas. La dysfonction de ce canal diminue le courant K+ sortant pendant la phase 3 du potentiel d’action, prolongeant ainsi sa durée.
Syndrome du QT long de type 3 (LQTS3)
Les patients atteints du LQTS3 ont un risque plus élevé de présenter des arythmies malignes au repos (sommeil) ou une bradycardie.15 La pénétrance de la mutation du gène SCN5A est de près de 90 %. L’ECG du LQTS3 montre généralement une onde T retardée et pointue et permet d’observer clairement l’allongement du segment ST12,13 (figure 2). Ces patients présentent généralement moins de symptômes que ceux atteints de LQTS1 ou LQTS2, mais les événements sont typiquement plus mortels.
Le gène affecté dans le LQTS3 est le SCN5A, qui code pour la sous-unité α du canal sodique Nav1.5 (Figure 1), situé sur le chromosome 3 (3p21-24) ; il est à l’origine de la maladie dans 5 à 10 % des cas. L’inactivation défectueuse du canal permet une entrée soutenue de Na+ pendant la phase 2 du potentiel d’action, prolongeant sa durée.
Syndrome du QT long de type 4 (LQTS4)
Le type 4 est une variété rare de LQTS, représentant près de 1% des cas. Il s’agit d’une forme atypique qui produit un large spectre d’arythmies, notamment une tachycardie ventriculaire polymorphe catécholaminergique, une fibrillation auriculaire, des altérations de la conduction intraventriculaire, un dysfonctionnement du nœud sinusal et une bradycardie6-18 ; en outre, le QTc peut se situer dans les limites normales chez de nombreux patients. Le gène affecté est ANKB, situé sur le chromosome 4 (4q25-27), qui code pour la synthèse de l’ankyrine-β, une protéine structurelle qui relie les protéines membranaires des cardiomyocytes aux protéines du cytosquelette. Ces protéines sont la pompe Na/K ATPase, l’échangeur Na/Ca et le récepteur de l’inositol triphosphate (InsP3R). Les mutations entraînant une perte de la fonction ankyrine-β conduisent à une augmentation de la concentration calcique intracellulaire et à des modifications de l’expression de la pompe N/K ATPase et de l’échangeur Na/Ca. La concentration élevée de calcium donne lieu à des post-dépolarisations précoces et retardées. Ainsi, les arythmies ventriculaires observées dans les mutations du gène ankyrin-β sont dues à des dépolarisations spontanées, généralement en réponse à une stimulation catécholaminergique.
Syndrome du QT long de type 5 (LQTS5)
Le type 5 trouve son origine dans des modifications de la séquence du gène KCNE1 situé sur le chromosome 21 (21q22.1p22.)19 KCNE1 code pour la synthèse de la sous-unité β du canal IKs, également appelée sous-unité minK, qui régule le canal IKs. Ce type représente moins de 1% des cas.
Syndrome du QT long de type 6 (LQTS6)
Le gène affecté dans le type 6 est KCNE2, situé sur le chromosome 21 (21q22.1).20 Ce gène code pour la sous-unité β du canal potassique, également connue sous le nom de sous-unité MiRP1, et il régule le canal IKr. Moins de 1 % des cas sont de type 6.
Syndrome du QT long de type 7 ou syndrome d’Andersen-Tawil (LQTS7)
Les résultats dysmorphiques et les altérations électrocardiographiques observés dans ce syndrome ont été décrits pour la première fois en 1971 par le Dr Andersen21 et revisités en 1994 par le Dr Tawil22, mais la description génétique/moléculaire n’a pas été rapportée avant 2001.23 Désormais connue sous le nom de syndrome d’Andersen-Tawil (ATS), cette affection est une altération autosomique dominante caractérisée par une paralysie périodique, un développement squelettique anormal, une arythmie ventriculaire du type impliquant de fréquentes extrasystoles ventriculaires, et une susceptibilité particulière à développer une fibrillation ventriculaire, notamment chez les femmes. Les altérations décrites dans le STA comprennent des extrasystoles ventriculaires (41 %), une tachycardie ventriculaire polymorphe non soutenue (23 %), une tachycardie ventriculaire bidirectionnelle (68 %) et une torsade de pointes (3 %).24 Parmi les caractéristiques dysmorphiques observées, citons une petite taille, une scoliose, une clinodactylie, un hypertélorisme, une implantation basse des oreilles, une micrognathie et un front large. L’expression de la maladie est variable, ce qui complique le diagnostic précoce.23,25 Les mutations du gène KCNJ2 situé sur le chromosome 17 (17q23), qui code pour la synthèse du canal potassique redresseur Kir 2.1, représentent 70 % des cas. Ce canal participe à la phase 4 du potentiel d’action. Plusieurs auteurs remettent en question l’inclusion de ce gène dans le groupe causal du LQTS, car l’intervalle QTc n’est que légèrement prolongé dans ce syndrome, voire normal, mais l’onde U est généralement proéminente, ce qui a conduit à une surestimation de l’intervalle QT. Le lecteur constatera que certains auteurs suggèrent que les mutations de KCNJ2 génèrent le SCA1 et non le LQTS7.24
Le syndrome du QT long de type 8 (LQTS8)
Le type 8 résulte de mutations du gène CACNA1 situé sur le chromosome 12 (12p13.3), qui code pour le canal calcique de type L Cav1.Il est à l’origine du syndrome de Timothy26, une affection caractérisée par des malformations cardiaques, un déficit immunologique intermittent, une hypoglycémie, des altérations cognitives dont l’autisme, une fusion interdigitale et un allongement de l’intervalle QT, qui entraîne une arythmie cardiaque et une mort subite27. Moins de 0,5% des cas sont de type 8.
Syndrome du QT long de type 9 (LQTS9)
Cette variété de LQTS se développe à partir de mutations dans le gène CAV3, situé sur le chromosome 3 (3p25), qui code pour la synthèse de la cavéoline 3. La cavéoline est une invagination de la membrane plasmique impliquée dans l’endocytose, l’homéostasie lipidique et la transduction du signal. Un composant important de cette structure est la cavéoline, qui a 3 sous-types connus ; le sous-type 3 est spécifique des muscles squelettiques et cardiaques. Certains canaux ioniques sont co-localisés dans la cavéole, notamment une isoforme cardiaque du canal sodique Nav1.5. Plusieurs mutations de cette protéine ont été récemment décrites. Celles-ci altèrent les propriétés biophysiques du canal sodique Nav1.5 in vitro, générant un phénotype similaire à celui observé dans le LQTS3.28 Moins de 1% des cas sont attribués à cette cause.
Syndrome du QT long de type 10 (LQTS10)
Le type 10 a été décrit dans un cas très sévère, avec un QTc >600 ms, une bradycardie fœtale et un bloc auriculo-ventriculaire (AV) 2:1. Il résulte de mutations dans le gène SCN4B, situé sur le chromosome 11 (11q23), qui code pour la sous-unité β4 du canal sodique. Quatre sous-types différents de sous-unités β ont été décrits, qui interagissent et régulent les diverses isoformes du canal sodique ; néanmoins, seul le sous-type 4 a été associé à l’arythmogenèse jusqu’à présent29. L’incidence des mutations de ce gène n’a pas été examinée, mais est estimée à
Mutations de la variété Jervell-Lange-Nielsen
Cette forme sévère de LQTS est causée par des mutations homozygotes30 ou hétérozygotes composées des gènes KCNQ1, et/ou KCNE1, qui codent pour le courant IKs ; c’est-à-dire une variété très sévère des formes LQTS1 ou LQTS5. Cette affection est caractéristiquement associée à une surdité congénitale. Les patients ont généralement un QTc>500 ms et des syncopes récurrentes, et présentent un risque élevé de mort subite. Les parents des patients atteints de cette variété sont généralement hétérozygotes et ont une maladie moins sévère, ou ne présentent aucun symptôme.31
DIAGNOSTIC DU SYNDROME DU LONG QT
Score de Schwartz
En 1985, Schwartz et al32 ont publié les critères de diagnostic du LQTS, qui ont été modifiés en 1993 et contiennent des directives importantes pour l’évaluation initiale des cas potentiels. Ce système utilise un score de 1 à 9 basé sur l’histoire familiale et les résultats cliniques et électrocardiographiques. La probabilité de la maladie est faible à un score ≥1, intermédiaire à 2-3, et élevée à ≥4 (tableau 2).

Diagnostic prénatal du syndrome du QT long
La bradycardie fœtale peut être l’une des premières manifestations cliniques du LQTS. Des séries rétrospectives ont montré que jusqu’à 70 % des patients chez qui on a diagnostiqué un STQL pendant l’enfance ont des antécédents de bradycardie, généralement accompagnée d’une anasarque fœtale.33 L’évaluation de la repolarisation cardiaque fœtale entre les semaines 14 et 39 est utile pour le diagnostic précoce du STQL.34
Le mosaïcisme gonadique pour le STQL a été associé à des pertes fœtales récurrentes pendant le troisième trimestre de la grossesse.35 Si la maladie est fortement suspectée, une amniocentèse après 16 semaines de gestation peut être utile pour établir le diagnostic, qui est facilement atteint lorsque l’un des parents est connu pour être porteur d’une mutation spécifique.36
ÉTUDE D’UN PATIENT AVEC SYNDROME DU LONG QT
Antécédents cliniques
Les antécédents familiaux et/ou personnels de mort subite sont d’une importance cruciale à la fois pour le diagnostic et la stratification du risque du LQTS. En outre, les facteurs précipitants et le contexte de la syncope peuvent indiquer le sous-type de LQTS. Lors de l’évaluation initiale d’un cas suspect, l’utilisation de médicaments pouvant prolonger l’intervalle QT doit être écartée.
Intervalle QT : Qu’est-ce qui est normal ?
L’intervalle QT doit être mesuré préférentiellement dans les dérivations II ou V5,37 où il a été prouvé qu’il avait une plus grande valeur prédictive.38 Cet intervalle indique la durée de la repolarisation ventriculaire et est mesuré du début de l’onde Q à la fin de l’onde T. Par convention, on utilise la formule proposée par Bazett39 pour corriger la durée de l’intervalle en fonction de la fréquence cardiaque (QTc=QT/√RR, exprimé en secondes). Bien que la mesure de l’intervalle QT semble simple, dans une étude multicentrique réalisée par Viskin et al40, moins de 40% des médecins autres que les cardiologues, moins de 50% des cardiologues et plus de 80% des spécialistes de l’arythmie savaient comment le mesurer correctement. Il est conseillé aux médecins de procéder à une mesure manuelle et de ne pas se fier aux mesures automatisées, qui peuvent être utiles pour d’autres intervalles, mais sont imprécises lorsqu’il s’agit de calculer l’intervalle QT. Le QT est un intervalle dynamique et les limites normales dépendent de plusieurs facteurs. Bien qu’un intervalle QTc de é440 ms chez l’homme et de é460 ms chez la femme soit considéré comme anormal, on peut trouver des porteurs de mutations ainsi que des individus sains dans cette fourchette (figure 3). Dans des familles avec LQTS1, Vincent et al41 ont démontré qu’aucun des cas avec un génotype positif n’avait un QTc470 ms. Monnig et al38 ont récemment montré que le QTc>440 ms suffit à détecter les patients présentant des mutations associées au LQTS, que le QTc>470 ms est utile pour identifier les patients risquant de développer des symptômes, et que le QTc>500 ms est retrouvé chez les patients symptomatiques sous traitement.
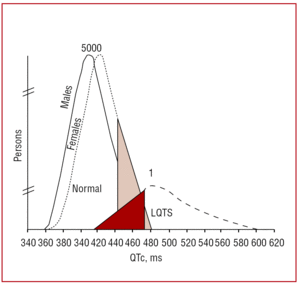
Figure 3. Modèle montrant la distribution de l’intervalle QT corrigé par la fréquence cardiaque (QTc) chez les patients présentant des mutations dans KVLQT1, HERG ou SCN5A, et chez les membres de leur famille non affectés. La courbe de gauche décrit la distribution des membres non affectés et la courbe de droite, les membres affectés.
Autres altérations électrocardiographiques associées au syndrome du QT long
Les patients atteints du LQTS peuvent présenter de multiples altérations de l’onde T : alternance de polarité, variations d’amplitude, encoche et aspect biphasique, entre autres42. L’alternance de l’onde T (figure 4A) est définie comme une variation battement par battement de l’amplitude, de la morphologie et de la polarité d’une onde T en rythme sinusal, sans variation du complexe QRS. Il s’agit d’un indicateur d’instabilité électrique43, reflétant la dispersion régionale de la repolarisation ventriculaire, et qui précède parfois la fibrillation ventriculaire44
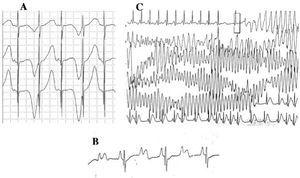
Figure 4. Altérations électrocardiographiques dans le syndrome du QT long. A : alternances électriques de l’onde T. B : bloc auriculo-ventriculaire 2:1. C : torsade de pointes autolimitée.
Les patients atteints du LQTS peuvent évoluer avec des signes de dysfonctionnement du nœud sinusal, de bradycardie et/ou de pauses.45 Les sous-types LQTS1 et LQTS3, en particulier ce dernier, présentent souvent une bradycardie sinusale,46 tandis que le LQTS4 a été associé à un dysfonctionnement du nœud sinusal.18
Depuis la décennie 1970-1980, la coexistence de défauts de conduction AV avec le LQTS47 a été observée (figure 4B). Le bloc AV deux à un est une manifestation peu fréquente et de mauvais pronostic qui peut être présente depuis le stade fœtal sous la forme d’une bradycardie persistante. L’incidence de cette anomalie a été rapportée à 4 %-5 %48 et elle est associée à une mortalité élevée malgré un traitement par bêta-bloquants et/ou stimulateurs cardiaques.49,50 Ce phénomène peut être expliqué par une longue durée du potentiel d’action. Lorsque la période réfractaire ventriculaire est prolongée, l’impulsion suivante de l’activité sinusale est bloquée car elle atteint les ventricules alors qu’ils sont encore en période réfractaire. Cette altération semble se produire exclusivement dans le LQTS, car la période réfractaire ventriculaire est plus longue que celle du système de conduction AV.51 La pente du complexe QRS est généralement abrupte et le bloc a été localisé dans la zone infrahis,46,51,52 mais le site du bloc peut dépendre du génotype. Jusqu’à présent, 4 gènes ont été associés au bloc 2:1 dans le LQTS : HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 et SCN4B (LQTS10).55
L’arythmie ventriculaire caractéristique du LQTS est connue sous le nom de torsade de pointes (figure 4C). Elle se présente lorsque l’intervalle QT est prolongé, quelle que soit l’étiologie. Il s’agit d’une tachycardie ventriculaire polymorphe due à une réentrée, caractérisée électrocardiographiquement par une torsion continue de l’axe QRS autour d’une ligne imaginaire. Elle est généralement précédée d’une pause suivie d’une extrasystole (intervalle RR court-long-court), comme le montre la figure.56-58 Elle peut aboutir à une fibrillation ventriculaire et à une mort subite. Si cela ne se produit pas, le patient peut seulement ressentir une syncope, et si l’épisode est bref, il peut passer inaperçu.
L’étude Holter
permet une évaluation complète et dynamique de l’intervalle QT. Occasionnellement, des épisodes spontanés d’arythmie ventriculaire asymptomatique sont enregistrés, ainsi que des épisodes de dysfonctionnement du nœud sinusal ou de bloc AV.
Epreuve d’effort
Les patients atteints du LQTS ne peuvent pas atteindre la fréquence cardiaque maximale attendue calculée en fonction de l’âge. De plus, à l’effort, l’intervalle QT peut présenter un comportement paradoxal, en augmentant plutôt qu’en diminuant.59,60 Le schéma électrocardiographique pendant l’épreuve d’effort sera différent selon le type de LQTS. Les patients atteints du LQTS1, en plus de ne pas atteindre la fréquence cardiaque maximale calculée pour leur âge, présentent fréquemment une augmentation de l’intervalle QT, tandis que ceux atteints du LQTS2 peuvent atteindre leur fréquence cardiaque prévue et ne présenter qu’une légère augmentation de l’intervalle QT, voire aucune.61,62 En général, les patients atteints du LQTS3 ont une réponse physiologique à l’exercice, c’est-à-dire un raccourcissement normal de l’intervalle QT.63 Les tests d’effort peuvent également être utiles pour évaluer la réponse au traitement et pour stratifier le risque dans les cas asymptomatiques, ou lorsqu’il y a des doutes quant aux événements conduisant à l’arythmie.
Dépistage génétique
Ces dernières années, les études génétiques du LQTS ont été limitées aux laboratoires de recherche. Néanmoins, les informations dérivées de ces efforts ont été extrêmement utiles pour traiter les patients, en particulier les cas à haut risque. La principale application du dépistage est peut-être le conseil génétique, mais il a également des implications importantes dans le traitement, qui peut être orienté en fonction de la voie affectée. La localisation précise d’une mutation donnée peut fournir des informations supplémentaires concernant l’évolution du risque. Les patients présentant des mutations dans la région transmembranaire de KCNQ1 (IKS) ont une plus grande probabilité de présenter des événements arythmiques que ceux présentant des mutations dans la région C-terminale64 ; il en va de même pour les patients présentant des mutations dans la région du pore de KCNH2 ou de HERG65 par rapport à ceux présentant des mutations dans les régions N- ou C-terminales66.
Le dépistage initial peut peut-être se limiter aux gènes KCNQ1, HERG et SCN5A, qui offrent la possibilité de rencontrer des mutations dans 65% des cas. Lorsque les résultats obtenus sont négatifs, le dépistage peut être étendu aux gènes KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 et SCN4B, ce qui augmentera la possibilité de résultats positifs de 5 % à 10 %.
Dépistage génétique post-mortem
Il est intéressant de constater que des mutations génétiques conduisant au LQTS ont été trouvées chez des enfants ayant subi une mort subite et dans des cas inexplicables de mort subite chez de jeunes adultes.
Des études génétiques post-mortem de patients présentant une mort subite et une autopsie négative ont montré des mutations conduisant au LQTS dans des pourcentages variables67-69 : près de 10 % chez les enfants et 35 % chez les jeunes adultes70-72. Sur la base de ces résultats, l’étude systématique de l’ECG a été proposée chez tous les nouveau-nés.73,74
L’étude génétique post-mortem, également connue dans la littérature sous le nom d' »autopsie moléculaire », outre les répercussions juridiques, a des implications importantes dans les familles qui pourraient être affectées sans qu’elles le sachent.
Polymorphismes régulateurs
Plusieurs polymorphismes fréquents ont été décrits dans la population LQTS, distribués dans presque tous les gènes associés à cette condition. Bien que ces modifications ne soient apparemment pas pathogènes, certaines peuvent avoir les effets suivants75-78:
1. Générer une susceptibilité individuelle à développer une arythmie.
2. Favoriser l’impact pathogène d’un autre changement non synonyme.
3. Diminuer l’effet pathogène d’un autre changement non synonyme.
C’est le cas du polymorphisme K897T de KCNH2 (HERG), qui est présent chez jusqu’à 15% de la population et qui est non seulement lié à la susceptibilité à certains médicaments,79 mais qui favorise également l’effet pathogène des mutations dans le même gène78. Un autre exemple est le polymorphisme S1103Y du gène SCN5A, présent principalement chez les Noirs, dont l’incidence est de près de 13 % et qui est associé à un risque accru de mort subite dans l’enfance80.
De manière intéressante, deux sites de traitement alternatifs générant deux types de canaux sodiques ont été décrits dans le produit du gène SCN5A, (qui code pour l’isoforme du canal sodique Nav1.5 chez l’homme) : l’un avec 2016 acides aminés contenant de la glutamine en position 1077 (Q1077), et l’autre avec 2015 acides aminés manquant de glutamine (Q1077del). Les transcrits de ces traitements alternatifs sont présents dans une proportion 2:1 dans un même cœur humain et plusieurs polymorphismes fréquents auront des effets différents sur le fonctionnement du canal, selon que le contexte est Q1077 ou Q1077del. Ceci a été initialement démontré avec le polymorphisme H558R de SCN5A, présent dans jusqu’à 30% de la population. Lorsque H558R a été exprimé dans le contexte de Q1077, une réduction profonde du courant ionique a été observée.81 Un effet similaire a été documenté avec le polymorphisme S524Y82. Ces résultats ont fourni des facteurs permettant d’expliquer la gravité variable de la maladie, ainsi que les différents phénotypes de la même mutation observés dans certaines familles.77
Test pharmacologique à l’adrénaline
Le test pharmacologique à l’adrénaline à faible dose est une option sûre et utile pour démasquer les cas suspects de LQTS avec un QTc limite. Il est particulièrement efficace pour détecter les formes asymptomatiques du LQTS1, avec une sensibilité de 92,5 %, une spécificité de 86 %, une valeur prédictive positive de 76 % et une valeur prédictive négative de 96 %. Il peut également être utile pour le diagnostic du LQTS2, avec une sensibilité et une spécificité plus faibles. Il n’est pas utile pour le LQTS3 ou d’autres formes de LQTS. Dans des conditions normales, la stimulation sympathique induit la phosphorylation du canal potassique IKs, optimisant sa fonction et donnant lieu à un raccourcissement du potentiel d’action. Chez les patients atteints de LQTS, en particulier de type 1, on observe une réponse paradoxale à l’administration d’adrénaline à faible dose (0,025-0,2 µg/kg/min) qui prolonge l’intervalle QT à plus de 30 ms83-86.
Prolongation de l’intervalle QT et TORSADE DE POINTES d’origine médicamenteuse
Une grande variété de médicaments utilisés dans différentes spécialités médicales peuvent provoquer une augmentation iatrogène de l’intervalle QT. Certains médicaments ont été retirés du marché en raison de cet effet indésirable (par exemple, l’astémizole et le cisapride, entre autres ; pour plus d’informations, consultez le site www.qtdrugs.org).87,88
L’arythmie ventriculaire secondaire à des médicaments non antiarythmiques survient chez moins d’un sujet exposé sur 10 000 à 100 000. Si l’on considère que les études cliniques comprennent entre 2 000 et 3 000 sujets, cet événement indésirable et fatal échapperait facilement à la détection pendant la phase clinique du développement des médicaments89. Ce point a suscité un énorme intérêt pour les aspects se référant à la sécurité dans l’étude et le développement de nouveaux médicaments.
Les facteurs liés à la susceptibilité individuelle comprennent le sexe féminin, l’hypocalcémie, l’hypomagnésémie, la bradycardie, l’insuffisance cardiaque, la postcardioversion, la fibrillation auriculaire, l’hypertrophie ventriculaire gauche, le LQTS non détecté, les polymorphismes prédisposants et les concentrations sériques élevées de médicaments prédisposants.90
Le canal qui interagit typiquement avec les médicaments est IKr, codé par le gène KCNH2(HERG), en raison de sa structure moléculaire. Les autres canaux potassiques ont 2 résidus proline inclinés vers le pore du canal, réduisant sa lumière. En revanche, IKr est dépourvu de ces résidus, ce qui génère un vestibule de pore plus large et facilite l’exposition aux grosses molécules. De plus, il possède 2 résidus aromatiques (tyrosine et phénylalanine) qui favorisent la liaison avec les molécules aromatiques présentes dans plusieurs médicaments capables de bloquer le canal.91
Comme il a été mentionné plus haut, la pénétrance du LQTS est incomplète et certains porteurs asymptomatiques de mutations pourraient manifester une arythmie maligne en recevant l’un de ces médicaments. De plus, des polymorphismes considérés comme fréquents dans la population confèrent une susceptibilité individuelle au développement de torsades de pointes lors de l’utilisation de certains médicaments. C’est le cas du polymorphisme R1047L, le deuxième plus fréquent dans le gène KCNH2, qui a été associé au développement de torsades de pointes lors de l’utilisation du médicament dofétilide.92 Au moins 20 polymorphismes du gène KCNH2 ont été décrits chez des personnes saines et leur effet sur la susceptibilité individuelle à développer des arythmies liées aux médicaments reste à déterminer.93 Les polymorphismes qui confèrent une susceptibilité au développement d’arythmies ventriculaires ont également été documentés dans le canal sodique Na1.5. C’est le cas du polymorphisme H558R, qui est présent chez jusqu’à 30 % de la population, ou S1103Y, qui est fréquent chez les Noirs80,81,90,94,95 ; Leur implication dans la susceptibilité induite par les médicaments n’a pas été étudiée.
SYNDROME DU QT LONG ET GROSSESSE
Le conseil génétique est important dans le cas du LQTS, mais en termes généraux, il n’y a pas de contre-indication à la grossesse chez les femmes qui sont porteuses, bien que chaque cas soit différent et doive être évalué individuellement dans le contexte approprié.
Il a été noté que le risque de présenter une arythmie ventriculaire maligne diminue avec la grossesse. En revanche, une plus grande vulnérabilité à présenter une arythmie maligne a été rapportée dans les 9 premiers mois après l’accouchement, en particulier chez les patients atteints de LQTS2. Ce risque diminue considérablement avec un traitement par bêta-bloquant.96
STRATIFICATION DU RISQUE
L’évolution du LQTS varie et est influencée par la durée de l’intervalle QTc, les facteurs environnementaux, l’âge, le génotype et la réponse au traitement.97,98 L’arythmie ventriculaire est plus fréquente dans le LQTS1 et le LQTS2, mais elle est plus sévère dans le LQTS3.99 Comme cela a été mentionné plus haut, les femmes sont particulièrement sensibles à l’arythmie maligne pendant la période post-partum.14
Le syndrome du QT long doit être considéré comme un risque élevé lorsqu’il est associé aux éléments suivants :
1. Surdité congénitale (syndrome de Jervell-Lange-Nielsen).
2. Syncope récurrente due à une tachyarythmie ventriculaire maligne.
3. Antécédents familiaux de mort subite.
4. QTc>500 ms.
5. Bloc auriculo-ventriculaire 2:1.
6. Alternance électrique de l’onde T.
7. Génotype LQTS3.
L’étude de Priori et al97 réalisée chez 647 patients a montré que la probabilité de présenter un événement majeur (syncope, arrêt cardiaque, mort subite) avant 40 ans est élevée (>50%) lorsque le QTc est >500 ms dans le LQTS1, le LQTS2 et chez les hommes avec le LQTS3. Une analyse du registre international du LQTS a récemment été rapportée. Le risque de mort subite a été analysé chez 2772 adolescents atteints de la maladie, et 3 facteurs associés à un risque plus élevé dans cette population ont été identifiés : QTc>530 ms, antécédents de syncope au cours des 10 dernières années et sexe ; les garçons de 10 à 12 ans avaient un risque plus élevé que les filles, mais dans la tranche d’âge de 13 à 20 ans, le risque était comparable.100
TRAITEMENT
Les patients symptomatiques qui ne reçoivent pas de traitement ont un taux de mortalité annuel de 20% et une mortalité à 10 ans de 50% après un premier événement d’arythmie ventriculaire. Bien qu’il soit clair qu’un traitement doit être mis en place en présence de symptômes, l’approche à utiliser chez les patients asymptomatiques fait encore l’objet de débats. Il a été documenté que l’arrêt cardiaque peut être la première manifestation de la maladie chez 9 % des patients,48 et que 12 % des patients asymptomatiques développeront des symptômes et pourront connaître une mort subite. Un traitement initial par bêta-bloquants doit être instauré chez tous les patients atteints de LQTS. La restriction de l’exercice est recommandable, mais les marqueurs de risque cliniques et électrocardiographiques constituent une base utile pour la prise de décision. Il est important d’informer les patients du risque lié à l’utilisation de plusieurs médicaments susceptibles de prolonger l’intervalle QT et de favoriser le développement d’une arythmie ventriculaire, comme cela a été mentionné plus haut. Le diagnostic génétique, outre qu’il permet un conseil familial approprié lié à la maladie, est une aide pour évaluer le pronostic et orienter le traitement spécifique.
Bêta-bloquants
Les bêta-bloquants sont le traitement de première intention pour le LQTS et tous les patients devraient les recevoir comme traitement initial.101 Ils permettent une réduction du risque d’événements cardiovasculaires allant jusqu’à 64 %100 et sont particulièrement efficaces chez les patients présentant des mutations du canal IKs (LQTS1),102 qui sont régulés en grande partie par le système sympathique. Les bêta-bloquants ne modifient pas l’intervalle QT, mais plutôt sa dispersion.103 Bien que ces médicaments diminuent l’incidence des événements,104,105 il a été démontré que 10 % des patients atteints du LQTS1, 23 % du LQTS2 et 32 % du LQTS3 présenteront des symptômes cardiovasculaires malgré le traitement.106 Les patients atteints du LQTS3, en particulier, ne semblent pas obtenir de bénéfices importants ; en fait, ce groupe de médicaments doit être utilisé avec prudence chez ces patients, car les épisodes d’arythmie ventriculaire dans le LQTS3 sont plus fréquents lorsque la fréquence cardiaque est faible. En termes généraux, 32 % des patients symptomatiques présenteront des symptômes récurrents au cours des 5 premières années avant de commencer un traitement par bêta-bloquant, et 14 % des patients sauvés d’un épisode de mort subite présenteront un autre événement similaire dans les 5 ans s’ils ne reçoivent que ce traitement107. Plusieurs bêtabloquants ont été utilisés dans le traitement du LQTS, principalement le nadolol (0,5-1 mg/kg/jour), le propranolol (2-4 mg/kg/jour), le métoprolol (0,5-1 mg/kg/jour) et l’aténolol (0,5-1 mg/kg/jour). Cependant, l’aténolol peut ne pas être bénéfique dans le cas du LQTS ; il a été notifié qu’au moins 75 % des patients qui ne répondaient pas au traitement par bêta-bloquant recevaient de l’aténolol, bien que cette constatation puisse être liée à l’utilisation de doses sous-optimales.104 Les tests d’effort sont utiles pour établir la dose appropriée. La fréquence cardiaque maximale ne doit pas dépasser 130 battements/min pendant le traitement.
Bloqueurs du canal sodique
Les mutations du canal sodique qui causent le LQTS3 produisent une inactivation défectueuse du canal ; le blocage du canal sodique s’est avéré utile chez ces patients. Des études réalisées avec la flécaïnide ont documenté des améliorations de la fréquence cardiaque, des altérations de l’onde T et de l’intervalle QT.108 On a également rapporté que la mexilétine améliorait les marqueurs de risque électrocardiographiques.63,109,110 Des études in vitro avec la ranolazine ont montré une diminution des effets délétères des mutations rapportées chez l’homme.111 Bien que les résultats soient encourageants, il faut garder à l’esprit qu’il n’existe pas d’études à long terme évaluant ce traitement, ni de résultats rapportés par de grandes séries. Les bloqueurs des canaux sodiques ne doivent pas être administrés en l’absence de diagnostic génétique confirmé.
Supplémentation en potassium et médicaments qui augmentent sa disponibilité
Les suppléments de potassium et/ou les médicaments d’épargne potassique, comme la spironolactone, raccourcissent l’intervalle QTc dans 24 % des cas112,113. Les médicaments qui favorisent l’ouverture des canaux potassiques, tels que l’aprikalim, le levcromakalim, le nicorandil et le pinacidil, se sont révélés utiles dans le traitement du LQTS. Les sous-types dans lesquels ils sont particulièrement bénéfiques sont le LQTS1 et le LQTS2.114
Stimulateurs et défibrillateurs
La stimulation par stimulateur cardiaque a été utilisée chez les patients présentant une arythmie dépendant d’une pause.115,116 Les patients atteints du LQTS3 bénéficient généralement davantage de ce traitement car la prévalence de la bradycardie est plus importante dans ce groupe. La stimulation DDD est indiquée chez les patients présentant une arythmie dépendant de la pause ou un bloc AV 2:1 de haut grade. Les fréquences programmées en dessous de 70 battements/min117 ne sont pas utiles pour prévenir l’arythmie ventriculaire. Il est recommandé de programmer le capteur en réponse rapide, car ces patients présentent généralement une accélération inappropriée de la fréquence cardiaque en réponse à l’exercice. Toutes les fonctions qui impliquent la présence de pauses doivent être désactivées, comme l’hystérésis et la fonction nocturne. La PARP (période réfractaire post-ventriculaire auriculaire) doit être aussi courte que possible. La fonction de régulation de la fréquence doit être activée pour prévenir la pause post-extrasystolique. Il ne faut pas oublier que la surdétection de l’onde T et les échecs de capture peuvent également donner lieu à des pauses. L’utilisation combinée d’un défibrillateur cardioverteur implantable (DCI) et de bêta-bloquants réduit considérablement l’incidence de la mort subite.118-120 L’indication de ces mesures est claire dans les cas à haut risque.121 La programmation de l’appareil varie en fonction des besoins de chaque patient, mais, en général, il faut éviter d’administrer un traitement lors d’événements asymptomatiques et autolimités ; à cette fin, un temps de détection de 15 s est indiqué. La tempête arythmique est une complication du traitement par AID. Près de 15 % des patients peuvent présenter cette complication, qui est due, en bonne partie, à une augmentation du tonus sympathique après le choc du DAI.118 Ce problème peut être géré en augmentant la dose de bêta-bloquant. Si cette mesure n’est pas utile, la résection des ganglions de la chaîne sympathique doit être envisagée.
Sympathectomie gauche
En 1971, la ganglionnectomie sympathique a été introduite comme une option thérapeutique utile chez ces patients.122 En 1991, Schwartz et al123 ont publié la première série de 85 patients ayant une mauvaise réponse au traitement par bêta-bloquants, chez qui une stellectomie gauche a été réalisée avec des résultats encourageants : un taux de survie à 5 ans de 94%. Actuellement, cette option thérapeutique est proposée aux patients à haut risque qui persistent à avoir des syncopes malgré un traitement par bêta-bloquant et/ou l’implantation d’un stimulateur cardiaque, et à ceux qui subissent des chocs fréquents de leur défibrillateur implanté. L’intervention consiste en une résection de la partie inférieure du ganglion stellaire et des ganglions thoraciques gauches T2 à T4 de la chaîne sympathique, la simple stellectomie gauche ne s’étant pas révélée suffisamment efficace. La thoracoscopie micro-invasive124,125 a été utilisée avec de bons résultats. La plus grande série de patients traités par cette méthode a été récemment rapportée et a montré une réduction significative du nombre d’épisodes de syncope ou de morts subites, ainsi qu’un taux de survie à 5 ans de 95 %. Chez les patients ayant déjà subi une syncope, la survie à 5 ans était de 97 %, avec une possibilité de récidive de 11 %, qui, dans la majorité des cas, consistait en un seul événement syncopal. On a également constaté une réduction significative du segment QT après une sympathectomie gauche. Malgré ces résultats favorables, la prévention de la mort subite n’est pas complète, mais a été réduite à 3 %. Chez les patients porteurs d’un DAI qui ont subi une intervention chirurgicale en raison de chocs multiples du défibrillateur, le nombre moyen d’événements est passé de 25 à 0, soit une réduction de 95 %. Un effet bénéfique a été confirmé dans le LQTS1. Les bénéfices sont susceptibles d’être plus faibles chez les patients atteints du LQTS2, et dans le LQTS3, son efficacité n’a pas été prouvée.126
Ablation
Il a été rapporté que l’ablation de l’extrasystole, qui dans certains cas initie l’arythmie ventriculaire, peut être réalisée avec une réduction de l’incidence des épisodes127. Cependant, il n’existe pas d’études à long terme avec un nombre approprié de patients pour justifier l’utilisation systématique de cette technique.
Voir l’éditorial aux pages 675-82
ABBREVIATIONS
AV : atrioventriculaire
AID : défibrillateur automatique implantable
ECG : électrocardiogramme
QTc : QT corrigé par la fréquence cardiaque
ATS : Syndrome d’Andersen-Tawil
LQTS : syndrome du QT long
Le Dr Medeiros reçoit un soutien économique du CONACyT et de FUNSALUD.