INDLEDNING
Langt QT-syndrom (LQTS) er karakteriseret ved en alvorligt ændret ventrikulær repolarisering, hvilket resulterer i en forlængelse af QT-intervallet på elektrokardiogrammet (EKG). Tilstanden prædisponerer patienterne for malign ventrikulær arytmi (torsade de pointes) og pludselig død. Den kliniske og elektrokardiografiske beskrivelse af langt QT-syndrom blev rapporteret i 1957 af Anton Jervell og Fred Lange Nielsen1 , som offentliggjorde deres undersøgelser af en familie af ikke-konsanguinerede forældre med 6 børn. Fire af børnene havde medfødt døvhed og synkopale episoder, og 3 præsenterede pludselig død. EKG-undersøgelse af disse patienter viste et usædvanligt langt QT-interval. Begge forældre var asymptomatiske, havde et normalt EKG og udviste ingen høreproblemer. I 1964 rapporterede Romano og Ward uafhængigt af hinanden et kardialt syndrom karakteriseret ved tilbagevendende synkoper, en familiehistorie med pludselig død og forlængelse af QT-intervallet uden neuronal døvhed.2 Senere genetiske undersøgelser viste, at det af Jervell og Lange Nielsen beskrevne syndrom, som ledsages af medfødt neuronal døvhed, svarer til homozygote mutationer, med en alvorlig fænotype og høj risiko for pludselig død. Den tilstand, der er kendt som Romano-Ward-syndromet, svarer generelt til heterozygote mutationer, patienterne udviser ikke hørelseforandringer, og sygdommens sværhedsgrad varierer betydeligt. Næsten et halvt århundrede senere, i 19953,4 , blev de vigtigste gener, der er forbundet med LQTS, beskrevet, og sygdommen blev anerkendt som en kardiel ionkanalforstyrrelse. Det var den første kardiale kanalopati, der blev beskrevet, og det er måske den mest udførligt undersøgte arytmogene ionkanalforstyrrelse til dato. Det kliniske billede varierer meget: patienten kan være asymptomatisk eller vise tilbagevendende synkope, kramper eller pludselig død som den første manifestation af sygdommen. Oprindeligt blev LQTS betragtet som et sjældent syndrom, og i realiteten er den alvorlige præsentation af sygdommen sporadisk. Ikke desto mindre anslås forekomsten af relaterede mutationer til 1/3000-5000 tilfælde,5 32 % af asymptomatiske bærere kan have et hjertefrekvenskorrigeret QT-interval (QTc) inden for de normale grænser, sygdommen overføres til 50 % af deres efterkommere, de er mere modtagelige for at udvikle arytmi sammenlignet med den generelle befolkning, og op til 20 % kan blive symptomatiske.6
Langt QT-syndrom udviser stor genetisk heterogenitet. Mere end 500 mutationer fordelt på 10 gener er blevet beskrevet i denne tilstand: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 og SCN4B. På trods af fremskridtene på dette område kan der ikke stilles en genetisk diagnose hos 25-30 % af patienterne.7,8 Sygdommens præsentation er hovedsagelig monogen6; polygene eller sammensatte varianter har normalt en mere alvorlig fænotype. Penetransen, dvs. de patienter, der har mutationen og manifesterer fænotypen, varierer fra 25 % til 90 %.9 Mindre hyppigt kan der være variationer i sygdommens ekspressivitet, med flere fænotyper som følge af den samme mutation. Molekylærgenetiske undersøgelser, der er udviklet i løbet af de sidste 11 år, har givet vigtige genotype-fænotypekorrelationer, som har været med til at styre behandlingstilgangen. Desuden er der gjort interessante observationer om den individuelle modtagelighed for arytmi i undersøgelser af de hyppige nonsynonyme polymorfismer i denne population, et aspekt, der har vakt stor interesse, især inden for farmakogenomikken.
KLASSIFIKATION AF LANGT QT SYNDROM
Generelle begreber
Den tidligere anvendte LQTS-klassifikation var baseret på homozygot eller heterozygot præsentation af sygdommen, hvilket giver anledning til henholdsvis Jervell-Lange-Nielsen syndrom (med døvhed) og Romano-Ward syndrom (uden døvhed). Den nuværende klassifikation lægger vægt på de genetiske fund, som illustreret i tabel 1. De 3 vigtigste gener, der er forbundet med sygdommen, blev beskrevet i 1995-1996. Disse gener, som koder for poredannende enheder af kaliumkanalerne IKs og IKr samt natriumkanalen Nav1.5, tegner sig for næsten 65 % af tilfældene. Selv om der i de efterfølgende år er blevet optaget yderligere syv gener på listen, tegner de sig kun for 5 % af tilfældene.
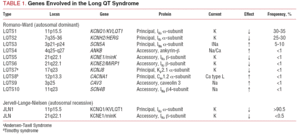
Ionkanaler er transmembranproteiner, der transporterer ioner gennem cellemembranen. De kanaler, der er involveret i LQTS, er selektive eller specialiserede i transport af en enkelt ion og er spændingsafhængige, dvs. at deres aktivering sker ved en specifik intracellulær spænding, som varierer alt efter kanalsubtypen. De elektriske og kontraktile fænomener, der forekommer i kardiomyocytten, styres af disse strukturer. Ionkanaler danner makromolekylære komplekser bestående af en hovedenhed, der danner kanalporen, og hjælpeproteiner, der regulerer den (figur 1). Den kanaldysfunktion, der ses i LQTS, kan forekomme på disse to steder: hovedproteinet eller de regulerende proteiner (tabel 1). Involvering af den poredannende enhed, kendt som alfa, genererer de tre mest almindelige undertyper af LQTS: LQTS1 (som påvirker IKs-kaliumkanalen), LQTS2 (som påvirker IKr-kaliumkanalen) og LQTS3 (som påvirker natriumkanalen). Da disse er de hyppigste undertyper, er de bedst karakteriseret klinisk og genetisk. Fænotype-genotypekorrelationerne i disse tre hovedformer er beskrevet i figur 2. På nuværende tidspunkt svarer Jervell-Lange-Nielsens syndrom til LQTS 1- og 5varianterne. Karakteristisk er, at disse patienter har medfødt døvhed og sammensatte homozygote eller heterozygote mutationer, der påvirker IKs strømmen. Romano-Ward syndromet omfatter varianter fra LQTS 1 til 10 og indebærer ikke døvhed.
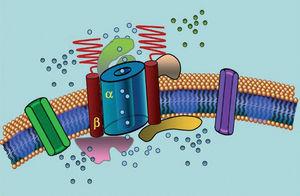
Figur 1. Skematisk fremstilling af det makromolekylære kompleks. Ionkanalerne er transmembranproteiner (α), der reguleres af forskellige proteiner; et af dem er den såkaldte β-underenhed.
Langt QT-syndrom type 1 (LQTS1)
Patienter med LQTS1 præsenterer normalt episoder af ventrikulær arytmi ved træning eller ved sympatisk stimulus (68 %).10 Svømning er blevet beskrevet som en sport, der udløser arytmi i LQTS1.11 Penetranscensen er næsten 62 % i denne undertype. T-bølgen hos disse patienter har ofte en bred base og meget langvarig varighed12,13 (Figur 2). Det er den hyppigste subtype og forklarer 30-35 % af tilfældene. Det berørte gen, KvLQT1 (eller KCNQ1), er placeret på kromosom 11 (11p15.5) og koder for IKs-kaliumkanalens α-underenhed. Aktionspotentialet forlænges ved en reduktion i den udgående K+-strøm under fase 3 af aktionspotentialet.
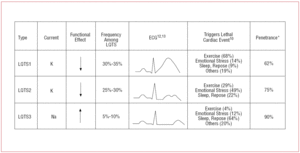
Figur 2. Genotype-fænotype korrelation i de mest hyppige lange QT-syndromer. *Henviser til tilfælde, der har mutationen og manifesterer fænotypen.
Langt QT-syndrom type 2 (LQTS2)
Patienter med LQTS2 har tendens til at præsentere ventrikulær arytmi som reaktion på følelsesmæssig stress (49 %) eller pludselige auditive stimuli (f.eks. et vækkeur) og mindre hyppigt under søvn (22 %) eller motion (29 %).10 Kvinder i postpartumperioden er særligt udsatte.14 Den estimerede penetrans er 79 %; derfor kan op til 20 % af tilfældene have et ikke-diagnostisk EKG. T-bølgen i LQTS2 er normalt lav-amplitude og bifid, med notching12,13 (Figur 2). Det berørte gen er KCNH2 eller HERG, der er placeret på kromosom 7 (7q35-36), som koder for IKr-kaliumkanalens α-underenhed, og som tegner sig for 25-30 % af tilfældene. Dysfunktion af denne kanal nedsætter den udgående K+-strøm i aktionspotentialets fase 3 og forlænger dets varighed.
Langt QT-syndrom type 3 (LQTS3)
Patienter med LQTS3 har en større risiko for at præsentere maligne arytmier under hvile (søvn) eller bradykardi.15 Penetranscensen af SCN5A-genmutationen er næsten 90 %. EKG’et i LQTS3 viser normalt en forsinket, spids T-bølge og giver mulighed for tydelig observation af ST-segmentforlængelsen12,13 (figur 2). Disse patienter har normalt færre symptomer end patienter med LQTS1 eller LQTS2, men hændelserne er karakteristisk set mere dødelige.
Det berørte gen i LQTS3 er SCN5A, som koder for Nav1.5-natriumkanalens α-underenhed (Figur 1), der er placeret på kromosom 3 (3p21-24); det er årsag til sygdommen i 5 %-10 % af tilfældene. Defekt inaktivering af kanalen tillader vedvarende input af Na+ under fase 2 af aktionspotentialet, hvilket forlænger dets varighed.
Langt QT-syndrom type 4 (LQTS4)
Type 4 er en sjælden variant af LQTS og udgør næsten 1 % af tilfældene. Det er en atypisk form, der giver et bredt spektrum af arytmier, herunder katekolaminerg polymorfisk ventrikulær takykardi, atrieflimren, intraventrikulære ledningsforandringer, sinusknude-dysfunktion og bradykardi6-18; desuden kan QTc være inden for de normale grænser hos mange patienter. Det berørte gen er ANKB, der er placeret på kromosom 4 (4q25-27), og som koder for syntese af ankyrin-β, et strukturelt protein, der forbinder kardiomyocytmembranproteiner med cytoskeletale proteiner. Disse proteiner er Na/K ATPase-pumpen, Na/Ca-udveksleren og inositoltrifosfatreceptoren (InsP3R). Mutationer, der medfører et tab af ankyrin-β-funktionen, fører til stigninger i den intracellulære calciumkoncentration og ændringer i ekspressionen af N/K ATPase og Na/Ca-udveksleren. Den forhøjede calciumkoncentration giver anledning til tidlige og forsinkede efterdepolarisationer. De ventrikulære arytmier, der observeres ved ankyrin-β-genmutationer, skyldes således spontane depolariseringer, som regel som reaktion på katekolaminerg stimulering.
Langt QT-syndrom type 5 (LQTS5)
Type 5 stammer fra ændringer i sekvensen af KCNE1-genet, der er placeret på kromosom 21 (21q22.1p22.)19 KCNE1 koder for syntese af IKs-kanalens β-underenhed, også kendt som minK-underenheden, som regulerer IKs-kanalen. Denne type tegner sig for mindre end 1 % af tilfældene.
Langt QT-syndrom type 6 (LQTS6)
Det berørte gen i type 6 er KCNE2, der er placeret på kromosom 21 (21q22.1).20 Dette gen koder for kaliumkanal β-underenheden, også kendt som MiRP1-underenheden, og det regulerer IKr-kanalen. Mindre end 1 % af tilfældene er type 6.
Langt QT-syndrom type 7 eller Andersen-Tawil syndrom (LQTS7)
De dysmorfiske fund og elektrokardiografiske ændringer, der ses i dette syndrom, blev først beskrevet i 1971 af Dr. Andersen21 og genbesøgt i 1994 af Dr. Tawil,22 men den genetiske/molekylære beskrivelse blev først rapporteret i 2001.23 Denne tilstand, der nu er kendt som Andersen-Tawil-syndromet (ATS), er en autosomalt dominerende ændring, der er karakteriseret ved periodisk lammelse, unormal skeletudvikling, ventrikulær arytmi af typen med hyppige ventrikulære ekstrasystoler og en særlig tilbøjelighed til at udvikle ventrikelflimmer, især hos kvinder. De forandringer, der er beskrevet i ATS, omfatter ventrikulære ekstrasystoler (41 %), ikke-vedvarende polymorfisk ventrikulær takykardi (23 %), bidirektionel ventrikulær takykardi (68 %) og torsade de pointes (3 %).24 Nogle af de observerede dysmorfiske karakteristika omfatter kort statur, skoliose, klinodaktyli, hypertelorisme, lav implantation af ørerne, mikrognathi og en bred pande. Sygdomsudtrykket varierer, hvilket vanskeliggør en tidlig diagnose.23,25 Mutationer i KCNJ2-genet, der er placeret på kromosom 17 (17q23), som koder for syntese af den rektificerende kaliumkanal Kir 2.1, tegner sig for 70 % af tilfældene. Denne kanal deltager i fase 4 af aktionspotentialet. Flere forfattere sætter spørgsmålstegn ved medtagelsen af dette gen i LQTS-årsagsgruppen, fordi QTc-intervallet kun er svagt forlænget i dette syndrom eller endog normalt, men U-bølgen er normalt fremtrædende, hvilket har ført til en overvurdering af QT-intervallet. Læseren vil opdage, at nogle forfattere antyder, at KCNJ2-mutationer genererer ATS1 og ikke LQTS7.24
Langt QT-syndrom type 8 (LQTS8)
Type 8 skyldes mutationer i CACNA1-genet, der er placeret på kromosom 12 (12p13.3), som koder for L-type calciumkanal Cav1.2. Den forårsager Timothy syndrom,26 en tilstand, der er karakteriseret ved kardiale misdannelser, intermitterende immunologisk mangel, hypoglykæmi, kognitive forandringer, herunder autisme, interdigital fusion og forlænget QT, som fører til hjerterytmeforstyrrelser og pludselig død27 . Mindre end 0,5 % af tilfældene er type 8.
Langt QT-syndrom type 9 (LQTS9)
Denne variant af LQTS udvikles på grund af mutationer i CAV3-genet, der er placeret på kromosom 3 (3p25), og som koder for caveolin 3-syntese. Caveola er en invagination af plasmamembranen, der er involveret i endocytose, lipidhomeostase og signaltransduktion. En vigtig komponent i denne struktur er caveolin, som har tre kendte subtyper; subtype 3 er specifik for skelet- og hjertemuskler. Nogle ionkanaler er samlokaliseret i caveola, herunder en kardiel isoform af natriumkanalen Nav1.5. Der er for nylig blevet beskrevet flere mutationer i dette protein. Disse ændrer de biofysiske egenskaber af natriumkanal Nav1.5 in vitro og genererer en fænotype, der ligner den, der er observeret i LQTS3.28 Mindre end 1 % af tilfældene tilskrives denne årsag.
Langt QT-syndrom type 10 (LQTS10)
Type 10 blev beskrevet i et meget alvorligt tilfælde med QTc >600 ms, føtal bradykardi og 2:1 atrioventrikulær (AV) blok. Den skyldes mutationer i SCN4B-genet, der er placeret på kromosom 11 (11q23), og som koder for natriumkanalens β4-underenhed. Der er beskrevet fire forskellige subtyper af β-subenheder, som interagerer og regulerer de forskellige natriumkanalisoformer; ikke desto mindre er kun subtype 4 hidtil blevet associeret med arytmogenese29 . Incidensen af mutationer i dette gen er ikke blevet undersøgt, men anslås til
Mutationer af Jervell-Lange-Nielsen-varianten
Denne alvorlige form for LQTS skyldes homozygote30 eller sammensatte heterozygote mutationer i KCNQ1- og/eller KCNE1-generne, som koder for IKs strømmen; dvs. en meget alvorlig variant af LQTS1- eller LQTS5-formerne. Denne tilstand er karakteristisk forbundet med medfødt døvhed. Patienterne har normalt en QTc>500 ms og tilbagevendende synkoper og har en høj risiko for pludselig død. Forældrene til patienter med denne variant er normalt heterozygote og har mindre alvorlig sygdom eller viser ingen symptomer.31
DIAGNOSE AF LANGT QT SYNDROM
Schwartz Score
I 1985 offentliggjorde Schwartz et al32 kriterierne for diagnosticering af LQTS, som blev ændret i 1993 og indeholder vigtige retningslinjer for den indledende vurdering af potentielle tilfælde. Dette system anvender en score fra 1 til 9 baseret på familiehistorien og de kliniske og elektrokardiografiske fund. Sandsynligheden for sygdom er lav ved en score på ≥1, mellemliggende ved 2-3 og høj ved ≥4 (tabel 2).

Prænatal diagnose af Long QT Syndrom
Fetal bradykardi kan være en af de første kliniske manifestationer af LQTS. Retrospektive serier har vist, at op til 70 % af patienter, der diagnosticeres med LQTS i barndommen, har en historie med bradykardi, som regel ledsaget af føtal hydrops.33 Vurdering af føtal hjerterepolarisering mellem uge 14 og 39 er nyttig til tidlig diagnose af LQTS.34
Gonadal mosaikisme for LQTS har været forbundet med tilbagevendende føtale tab i løbet af tredje trimester af graviditeten.35 Hvis sygdommen er stærkt mistænkt, kan fostervandsprøve efter 16 ugers graviditet være nyttig til at fastslå diagnosen, som let nås, når en af forældrene er kendt som bærer af en specifik mutation.36
UDVIDELSE AF EN PATIENT MED LANGT QT SYNDROM
Klinisk anamnese
En familie- og/eller personlig historie med pludselig død er af afgørende betydning for både diagnosen og risikostratificeringen af LQTS. Desuden kan udløsende faktorer og konteksten for synkope indikere LQTS-subtypen. Ved den indledende vurdering af et mistænkt tilfælde bør brugen af lægemidler, der kan forlænge QT-intervallet, udelukkes.
QT-interval: Hvad er normalt?
QT-intervallet bør fortrinsvis måles i afledning II eller V5,37 hvor det har vist sig at have større prædiktiv værdi.38 Dette interval angiver varigheden af ventrikulær repolarisering og måles fra begyndelsen af Q-bølgen til slutningen af T-bølgen. Konventionelt anvendes den formel, der er foreslået af Bazett39 , til at korrigere varigheden af intervallet i forhold til hjertefrekvensen (QTc=QT/√RR, udtrykt i sekunder). Selv om måling af QT-intervallet synes enkel, vidste mindre end 40 % af andre læger end kardiologer, mindre end 50 % af kardiologerne og mere end 80 % af specialisterne i arytmi i en multicenterundersøgelse udført af Viskin et al40 , hvordan QT-intervallet skal måles korrekt. Det er tilrådeligt for læger at foretage manuel måling og ikke stole på automatiserede målinger, som kan være nyttige for andre intervaller, men som er upræcise ved beregning af QT-intervallet. QT er et dynamisk interval, og de normale grænser afhænger af flere faktorer. Selv om et QTc-interval på é440 ms hos mænd og é460 ms hos kvinder betragtes som unormalt, kan man finde bærere af mutationer såvel som raske personer inden for dette interval (figur 3). I familier med LQTS1 viste Vincent et al41 , at ingen af tilfældene med en positiv genotype havde et QTc470 ms. Monnig et al38 viste for nylig, at QTc>440 ms er tilstrækkeligt til at opdage patienter med LQTS-associerede mutationer, QTc>470 ms er nyttigt til at identificere patienter i risiko for at udvikle symptomer, og QTc>500 ms findes hos symptomatiske patienter under behandling.
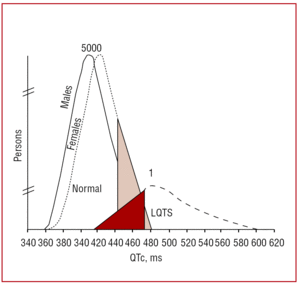
Figur 3. Model, der viser fordelingen af det hjertefrekvenskorrigerede QT-interval (QTc) hos patienter med mutationer i KVLQT1, HERG eller SCN5A og deres upåvirkede familiemedlemmer. Kurven til venstre beskriver fordelingen af ikke-påvirkede medlemmer og kurven til højre, påvirkede medlemmer.
Andre elektrokardiografiske ændringer forbundet med langt QT-syndrom
Patienter med LQTS kan præsentere flere T-bølgeændringer: polaritetsalteraner, amplitudevariationer, notching og et bifasisk udseende, blandt andre.42 T-bølge alternans (Figur 4A) er defineret som en slag-til-slag variation i amplitude, morfologi og polaritet af en sinusrytme T-bølge uden variationer i QRS-komplekset. Den er en indikator for elektrisk ustabilitet,43 der afspejler regional spredning af ventrikulær repolarisering og går lejlighedsvis forud for ventrikelflimmer.44
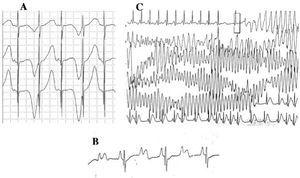
Figur 4. Elektrokardiografiske ændringer ved langt QT-syndrom. A: T-bølge elektrisk alternans. B: atrioventrikulær blok 2:1. C: selvbegrænsede torsade de pointes.
Patienter med LQTS kan udvikle sig med tegn på sinusknude-dysfunktion, bradykardi og/eller pauser.45 LQTS1- og LQTS3-subtyperne, især sidstnævnte, udviser ofte sinusbradykardi,46 mens LQTS4 er blevet associeret med sinusknude-dysfunktion.18
Siden årtiet 1970-1980 er der blevet observeret sameksistens af AV-ledningsdefekter med LQTS47 (Figur 4B). To-til-en AV-blok er en sjældent forekommende manifestation med en dårlig prognose, der kan være til stede siden fosterstadiet i form af vedvarende bradykardi. Forekomsten af denne abnormitet er rapporteret til 4-5 %48 , og den er forbundet med høj mortalitet på trods af behandling med betablokkere og/eller pacemakere.49,50 Dette fænomen kan forklares ved en langvarig varighed af aktionspotentialet. Når den ventrikulære refraktære periode er forlænget, blokeres den efterfølgende impuls fra sinusaktiviteten, fordi den når ventriklerne, mens de stadig befinder sig i den refraktære periode. Denne ændring synes udelukkende at forekomme i LQTS, fordi den ventrikulære refraktære periode er længere end AV-ledningssystemets.51 QRS-kompleksets hældning er normalt stejl, og blokaden er blevet lokaliseret i infraHis-området,46,51,52 men stedet for blokaden kan afhænge af genotypen. Indtil nu er 4 gener blevet relateret til 2:1 blokering i LQTS: HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 og SCN4B (LQTS10).55
Den karakteristiske ventrikulære arytmi for LQTS er kendt som torsade de pointes (Figur 4C). Den opstår, når QT-intervallet er forlænget, uanset ætiologien. Det er en polymorf ventrikulær takykardi som følge af reentry, der elektrokardiografisk er karakteriseret ved en kontinuerlig vridning af QRS aksen omkring en imaginær linje. Den indledes almindeligvis med en pause efterfulgt af en ekstrasystoli (kort-langt-kort RR-interval), som vist i figuren.56-58 Den kan kulminere i ventrikelflimmer og pludselig død. Hvis dette ikke sker, oplever patienten måske kun synkope, og hvis episoden er kortvarig, kan den gå uopdaget hen.
Holter
Holterundersøgelse giver en fuldstændig, dynamisk vurdering af QT-intervallet. Lejlighedsvis registreres spontane episoder af asymptomatisk ventrikulær arytmi samt episoder af sinusknude-dysfunktion eller AV-blok.
Motion Stress Test
Patienter med LQTS kan ikke nå den maksimale forventede hjertefrekvens beregnet i forhold til alder. Desuden kan QT-intervallet under anstrengelse udvise paradoksal adfærd ved at stige i stedet for at falde.59,60 Det elektrokardiografiske mønster under anstrengelsestestest vil være forskelligt afhængigt af typen af LQTS. Patienter med LQTS1 viser ud over ikke at nå den maksimale beregnede puls for deres alder ofte et øget QT-interval, mens patienter med LQTS2 kan nå deres forventede puls og kun vise en mild stigning i QT-intervallet eller slet ingen.61,62 Generelt har patienter med LQTS3 et fysiologisk respons på anstrengelse, dvs. en normal forkortelse af QT-intervallet.63 Stresstestning kan også være nyttig til vurdering af behandlingsrespons og til stratificering af risiko i asymptomatiske tilfælde, eller når der er tvivl om de begivenheder, der fører til arytmi.
Genetisk screening
I de seneste år har genetiske undersøgelser af LQTS været begrænset til forskningslaboratorier. Ikke desto mindre har de oplysninger, der stammer fra disse bestræbelser, været yderst nyttige for behandlingen af patienter, især højrisikofælde. Den vigtigste anvendelse af screening er måske i forbindelse med genetisk rådgivning, men den har også vigtige konsekvenser for behandlingen, som kan orienteres efter den berørte kanal. Den præcise placering af en given mutation kan give yderligere oplysninger om udviklingen af risikoen. Patienter med mutationer i transmembranregionen af KCNQ1 (IKS) har større sandsynlighed for at få arytmiske hændelser end patienter med mutationer i den C-terminale region64; det samme gælder for patienter med mutationer i poreregionen af KCNH2 eller HERG65 sammenlignet med patienter med mutationer i den N- eller C-terminale region66 .
Den indledende screening kan måske begrænses til KCNQ1-, HERG- og SCN5A-generne, som giver mulighed for at støde på mutationer i 65 % af tilfældene. Når de opnåede resultater er negative, kan screeningen udvides til KCNE1-, KCNE2-, ANKB-, KCNJ2-, CACNA1-, CAV3- og SCN4B-generne, hvilket vil øge muligheden for positive resultater med 5 % til 10 %.
Postmortem genetisk screening
Det er interessant, at genmutationer, der fører til LQTS, er blevet fundet hos børn, der har oplevet pludselig død, og i uforklarlige tilfælde af pludselig død hos unge voksne.
Postmortale genetiske undersøgelser af patienter med pludselig død og negativ obduktion har vist mutationer, der fører til LQTS, i varierende procentdele67-69: tæt på 10 % hos børn og 35 % hos unge voksne70-72 . På baggrund af disse resultater er der blevet foreslået rutinemæssige EKG-undersøgelser hos alle nyfødte.73,74
Postmortale genetiske undersøgelser, også kendt i litteraturen som “molekylær obduktion”, har ud over de juridiske konsekvenser vigtige implikationer i familier, der kan være påvirket, uden at de ved det.
Regulatoriske polymorfismer
Der er beskrevet flere hyppigt forekommende polymorfismer i LQTS-populationen, fordelt på næsten alle gener, der er forbundet med denne tilstand. Selv om disse ændringer tilsyneladende ikke er patogene, kan nogle af dem have følgende virkninger75-78:
1. Generere individuel modtagelighed for at udvikle arytmi.
2. 2. Begynder den patogene virkning af en anden ikke-synonym ændring.
3. Mindsker den patogene virkning af en anden ikke-synonym ændring.
Dette er tilfældet med K897T-polymorfismen i KCNH2 (HERG), som findes hos op til 15 % af befolkningen og ikke kun er forbundet med modtagelighed for visse lægemidler,79 men også begunstiger den patogene virkning af mutationer i samme gen78. Et andet eksempel er S1103Y-polymorfismen i SCN5A-genet, som hovedsagelig findes hos sorte, og som har en forekomst på næsten 13 % og er forbundet med en øget risiko for pludselig død i barndommen80 .
Interessant nok er der beskrevet to alternative behandlingssteder, der genererer to typer natriumkanaler, i produktet af SCN5A-genet (som koder for Nav1.5-natriumkanalisoformen hos mennesker): et med 2016 aminosyrer, der indeholder glutamin i 1077-positionen (Q1077), og et andet med 2015 aminosyrer, der mangler glutamin (Q1077del). Transskriptioner af disse alternative bearbejdninger er til stede i et 2:1-forhold i det samme menneskehjerte, og flere hyppige polymorfismer vil have forskellige virkninger på kanalens funktion, afhængigt af om konteksten er Q1077 eller Q1077del. Dette blev oprindeligt vist med H558R-polymorfismen i SCN5A, som findes hos op til 30 % af befolkningen. Når H558R blev udtrykt i forbindelse med Q1077, blev der observeret en dybtgående reduktion i ionstrømmen.81 En lignende effekt blev dokumenteret med S524Y82-polymorfi. Disse fund har givet faktorer til at forklare sygdommens varierende sværhedsgrad samt de forskellige fænotyper af den samme mutation, der er observeret i nogle familier.77
Farmakologisk testning med adrenalin
Farmakologisk testning med lavdosis adrenalin er en sikker og nyttig mulighed for at afmaske mistænkte tilfælde af LQTS med en borderline QTc. Den er særlig effektiv til påvisning af asymptomatiske former for LQTS1 med en sensitivitet på 92,5 %, en specificitet på 86 %, en positiv prædiktiv værdi på 76 % og en negativ prædiktiv værdi på 96 %. Den kan også være nyttig til diagnosticering af LQTS2, men med lavere sensitivitet og specificitet. Den er ikke nyttig i forbindelse med LQTS3 eller andre former for LQTS. Under normale forhold inducerer sympatisk stimulering fosforylering af IKs-kaliumkanalen, hvilket optimerer dens funktion og giver anledning til en forkortelse af aktionspotentialet. Hos patienter med LQTS, især type 1, observeres et paradoksalt respons på indgivelse af lavdosis adrenalin (0,025-0,2 µg/kg/min), der forlænger QT-intervallet til mere end 30 ms83-86.
QT-intervalforlængelse og lægemiddelinduceret TORSADE DE POINTES
En lang række lægemidler, der anvendes inden for forskellige medicinske specialer, kan forårsage en iatrogen forlængelse af QT-intervallet. Nogle lægemidler er blevet fjernet fra markedet på grund af denne uønskede virkning (f.eks. astemizol og cisaprid, blandt andre; for yderligere oplysninger, se www.qtdrugs.org).87,88
Ventrikulær arytmi sekundært til ikke-antiarytmiske lægemidler forekommer hos mindre end én ud af hver 10 000 til 100 000 eksponerede personer. I betragtning af at kliniske undersøgelser omfatter mellem 2 000 og 3 000 forsøgspersoner, ville denne uønskede og fatale bivirkning let kunne undgå at blive opdaget i den kliniske fase af lægemiddeludviklingen89 . Dette punkt har skabt enorm interesse for aspekter, der henviser til sikkerhed i forbindelse med undersøgelse og udvikling af nye lægemidler.
Faktorerne i forbindelse med individuel modtagelighed omfatter kvindeligt køn, hypokalcæmi, hypomagnesiæmi, bradykardi, hjertesvigt, postkardioversion, atrieflimren, venstre ventrikulær hypertrofi, uopdaget LQTS, prædisponerende polymorfismer og høje serumkoncentrationer af prædisponerende lægemidler.90
Den kanal, der typisk interagerer med lægemidler, er IKr, der er kodet af KCNH2(HERG)-genet, på grund af sin molekylære struktur. Andre kaliumkanaler har 2 prolinrester vinklet mod kanalporen, hvilket reducerer dens lumen. I modsætning hertil mangler IKr disse rester, der dannes en større porevestibule, og eksponering for store molekyler lettes. Desuden har den 2 aromatiske rester (tyrosin og phenylalanin), der begunstiger binding med aromatiske molekyler, der er til stede i flere lægemidler, som er i stand til at blokere kanalen.91
Som nævnt ovenfor er LQTS-penetransen ufuldstændig, og nogle asymptomatiske mutationsbærere kan manifestere malign arytmi ved at få et af disse lægemidler. Desuden giver polymorfismer, der anses for hyppige i befolkningen, individuel modtagelighed for udvikling af torsade de pointes, når nogle lægemidler anvendes. Dette er tilfældet med R1047L-polymorfismen, den næsthyppigste i KCNH2, som er blevet forbundet med udvikling af torsade de pointes ved brug af lægemidlet dofetilid.92 Mindst 20 polymorfismer i KCNH2-genet er blevet beskrevet hos raske personer, og deres virkning på den individuelle modtagelighed for udvikling af lægemiddelrelateret arytmi er endnu ikke fastlagt.93 Polymorfismer, der giver modtagelighed for udvikling af ventrikulær arytmi, er også blevet dokumenteret i natriumkanalen Na1.5. Det gælder H558R-polymorfismen, som findes hos op til 30 % af befolkningen, eller S1103Y, som er hyppig hos sorte80,81,90,94,95; deres betydning for lægemiddelinduceret modtagelighed er ikke blevet undersøgt.
LANGT QT SYNDROM OG GRAVNING
Genetisk rådgivning er vigtig i forbindelse med LQTS, men generelt er der ingen kontraindikation for graviditet hos kvinder, der er bærere, selv om hvert enkelt tilfælde er forskelligt og bør vurderes individuelt i den relevante sammenhæng.
Det er blevet bemærket, at risikoen for at få maligne ventrikulære arytmier falder med graviditet. Derimod er der rapporteret om større sårbarhed for at præsentere malign arytmi inden for de første 9 måneder efter fødslen, især hos patienter med LQTS2. Denne risiko falder betydeligt ved betablokkerbehandling.96
RISK STRATIFIKATION
Udviklingen af LQTS varierer og påvirkes af QTc-intervallets varighed, miljøfaktorer, alder, genotype og respons på behandling.97,98 Ventrikulær arytmi er hyppigere i LQTS1 og LQTS2, men er mere alvorlig i LQTS3.99 Som nævnt ovenfor er kvinder særligt modtagelige for maligne arytmier i den postpartale periode.14
Langt QT-syndrom bør betragtes som højrisiko, når det er forbundet med følgende:
1. Medfødt døvhed (Jervell-Lange-Nielsens syndrom).
2. 2. Recidiverende synkope på grund af malign ventrikulær takyarytmi.
3. Familiehistorie med pludselig død.
4. QTc>500 ms.
5. 2:1 atrioventrikulær blok.
6. T-bølge elektrisk alternans.
7. LQTS3-genotype.
Undersøgelsen af Priori et al97 udført på 647 patienter viste, at sandsynligheden for at få en større hændelse (synkope, hjertestop, pludselig død) før 40-årsalderen er høj (>50 %), når QTc er >500 ms hos LQTS1, LQTS2 og hos mænd med LQTS3. For nylig blev der rapporteret en analyse af det internationale LQTS-register. Risikoen for pludselig død blev analyseret hos 2772 unge med sygdommen, og der blev identificeret 3 faktorer, der er forbundet med højere risiko i denne population: QTc>530 ms, historie med synkope inden for de seneste 10 år og køn; 10-12-årige drenge havde en højere risiko end piger, men i aldersgruppen 13-20 år var risikoen sammenlignelig.100
BEHANDLING
Symptomatiske patienter, der ikke modtager behandling, har en årlig dødelighed på 20 % og en 10-årig dødelighed på 50 % efter en første hændelse med ventrikulær arytmi. Selv om det er klart, at der bør iværksættes behandling, når der er symptomer, er det stadig omdiskuteret, hvilken fremgangsmåde der skal anvendes hos asymptomatiske patienter. Det er blevet dokumenteret, at hjertestop kan være den første manifestation af sygdommen hos 9 % af patienterne48 , og at 12 % af asymptomatiske patienter vil udvikle symptomer og kan opleve pludselig død. Initial behandling med betablokkere bør påbegyndes hos alle patienter med LQTS. Træningsbegrænsning kan anbefales, men de kliniske og elektrokardiografiske risikomarkører er et nyttigt grundlag for beslutningstagning. Det er vigtigt at informere patienterne om risikoen ved at bruge flere lægemidler, der kan forlænge QT-intervallet og fremme udviklingen af ventrikulær arytmi, som nævnt ovenfor. Genetisk diagnose er, ud over at give mulighed for passende familierådgivning i forbindelse med sygdommen, en hjælp til at vurdere prognosen og orientere specifik behandling.
Beta-blokkere
Beta-blokkere er førstevalgsbehandlingen af LQTS, og alle patienter bør modtage dem som den indledende behandling.101 De giver en reduktion i risikoen for kardiovaskulære hændelser på op til 64 %100 og er særligt effektive hos patienter med IKs-kanalmutationer (LQTS1)102 , som i høj grad reguleres af det sympatiske system. Betablokkere ændrer ikke QT-intervallet, men derimod dets spredning.103 Selv om disse lægemidler mindsker forekomsten af hændelser,104,105 har det vist sig, at 10 % af patienterne med LQTS1, 23 % med LQTS2 og 32 % med LQTS3 vil få kardiovaskulære symptomer på trods af behandlingen.106 Især patienter med LQTS3 synes ikke at opnå væsentlige fordele; faktisk bør denne lægemiddelgruppe anvendes med forsigtighed hos disse patienter, fordi episoder af ventrikulær arytmi hos LQTS3 er mere almindelige, når hjertefrekvensen er lav. Generelt set vil 32 % af de symptomatiske patienter få tilbagevendende symptomer inden for de første 5 år, før de påbegynder betablokkerbehandling, og 14 % af de patienter, der er reddet fra en episode med pludselig død, vil få en anden lignende hændelse inden for 5 år, hvis de kun får denne behandling107 . Der er blevet anvendt flere betablokkere til behandling af LQTS, primært nadolol (0,5-1 mg/kg/dag), propranolol (2-4 mg/kg/dag), metoprolol (0,5-1 mg/kg/dag) og atenolol (0,5-1 mg/kg/dag). Atenolol er dog muligvis ikke gavnligt ved LQTS; det er blevet meddelt, at mindst 75 % af de patienter, der ikke reagerede på betablokkerbehandling, fik atenololol, selv om dette resultat kan være relateret til brugen af suboptimale doser.104 Træningstest er nyttig til at fastlægge den passende dosis. Den maksimale hjertefrekvens bør ikke overstige 130 slag/min under behandlingen.
Natriumkanalblokkere
Natriumkanalmutationer, der forårsager LQTS3, giver defekt inaktivering af kanalen; natriumkanalblokering har vist sig at være nyttig hos disse patienter. Undersøgelser udført med flecainid har dokumenteret forbedringer i hjertefrekvens, T-bølgeforandringer og QT-interval.108 Mexiletin er også blevet rapporteret til at forbedre de elektrokardiografiske risikomarkører.63,109,110 In vitro-undersøgelser med ranolazin har vist fald i de skadelige virkninger af mutationer rapporteret hos mennesker.111 Selv om resultaterne er opmuntrende, skal man huske på, at der ikke findes nogen langtidsundersøgelser, der vurderer denne behandling, og der er ikke rapporteret resultater fra store serier. Natriumkanalblokkere bør ikke administreres, hvis der ikke foreligger en bekræftet genetisk diagnose.
Kaliumtilskud og lægemidler, der øger dets tilgængelighed
Kaliumtilskud og/eller kaliumbesparende lægemidler, såsom spironolacton, forkorter QTc-intervallet i 24 % af tilfældene.112,113 Lægemidler, der begunstiger åbning af kaliumkanalerne, såsom aprikalim, levcromakalim, nicorandil og pinacidil, har vist sig at være nyttige i behandlingen af LQTS. De undertyper, hvor de er særligt gavnlige, er LQTS1 og LQTS2.114
Pacemakere og defibrillatorer
Pacemakerstimulering er blevet anvendt til patienter med pauseafhængig arytmi.115,116 Patienter med LQTS3 har normalt mere gavn af denne behandling, fordi prævalensen af bradykardi er større i denne gruppe. DDD-stimulering er indiceret hos patienter med pauseafhængig arytmi eller højgrads 2:1 AV-blokering. Frekvenser, der er programmeret til under 70 slag/min117 , er ikke nyttige til forebyggelse af ventrikulær arytmi. Det anbefales at programmere sensoren til hurtig respons, da disse patienter normalt har en uhensigtsmæssig hjertefrekvensacceleration som reaktion på træning. Alle funktioner, der indebærer tilstedeværelsen af pauser, bør slås fra, f.eks. hysterese- og natfunktion. PARP (postventrikulær atriel refraktær periode) skal være så kort som muligt. Frekvensreguleringsfunktionen bør være aktiveret for at forhindre postextrasystolisk pause. Det skal huskes, at overfølsomhed af T-bølger og fejl i opfangningen også kan give anledning til pauser. Kombineret brug af en implantabel cardioverter-defibrillator (ICD) og betablokkere mindsker forekomsten af pludselig død betydeligt.118-120 Indikationen for disse foranstaltninger er klar i højrisikofælde.121 Programmering af apparatet vil variere alt efter den enkelte patients behov, men generelt bør administration af behandling ved asymptomatiske, selvbegrænsede hændelser undgås; til dette formål er en detektionstid på 15 s indiceret. Arytmisk storm er en komplikation ved AID-behandling. Næsten 15 % af patienterne kan opleve denne komplikation, som i høj grad skyldes den øgede sympatiske tone efter ICD-chokket.118 Dette problem kan håndteres ved at øge betablokkerdosis. Hvis denne foranstaltning ikke er nyttig, bør resektion af sympatikkædens ganglier overvejes.
Venstre sympathektomi
I 1971 blev sympatisk gangliektomi introduceret som en nyttig terapeutisk mulighed hos disse patienter.122 I 1991 offentliggjorde Schwartz et al123 den første serie af 85 patienter med dårligt respons på betablokkerbehandling, hos hvem der blev foretaget en venstre stellektomi med opmuntrende resultater: en 5-års overlevelsesrate på 94%. I øjeblikket tilbydes denne behandlingsmulighed til højrisikopatienter, der fortsat har synkope på trods af betablokkerbehandling og/eller pacemakerimplantation, og til patienter, der hyppigt får stød fra deres implanterede defibrillator. Proceduren består i resektion af den inferior del af stellatganglien og de venstre thorakale ganglier T2 til T4 i den sympatiske kæde, da simpel venstre stellektomi ikke har vist sig at være tilstrækkelig effektiv. Mikroinvasiv thorakoskopi124,125 er blevet anvendt med gode resultater. Den største serie af patienter, der er behandlet med denne metode, blev for nylig rapporteret og viste en betydelig reduktion i antallet af synkopeepisoder eller pludselige dødsfald samt en 5-års overlevelsesrate på 95 %. Hos patienter med tidligere synkoper var 5-årsoverlevelsen 97 % med 11 % mulighed for recidiv, som i de fleste tilfælde bestod af en enkelt synkopebegivenhed. Der var også en betydelig reduktion af QT-segmentet efter venstre sympatektomi. På trods af disse gunstige resultater er forebyggelsen af pludselig død ikke fuldstændig, men er blevet reduceret til 3 %. Hos patienter med en ICD, der blev opereret på grund af flere defibrillatorchok, faldt det gennemsnitlige antal hændelser fra 25 til 0, hvilket svarer til en reduktion på 95 %. En gavnlig effekt blev bekræftet i LQTS1. Fordelene er sandsynligvis mindre hos patienter med LQTS2, og hos LQTS3 er effektiviteten ikke påvist.126
Ablation
Det er blevet rapporteret, at ablation af den ekstrasystole, som i nogle tilfælde initierer den ventrikulære arytmi, kan udføres med en reduktion i forekomsten af episoder.127 Der findes dog ingen langtidsundersøgelser med et passende antal patienter, der kan retfærdiggøre rutinemæssig anvendelse af denne teknik.
Se editorial på side 675-82
FORKORTELSER
AV: atrioventrikulær
AID: automatisk implantabel defibrillator
ECG: elektrokardiogram
QTc: hjertefrekvenskorrigeret QT
ATS: Andersen-Tawil-syndrom
LQTS: langt QT-syndrom
Dr. Medeiros modtager økonomisk støtte fra CONACyT og FUNSALUD.