Klassifikation von AML und CML
Die französisch-amerikanisch-britische (FAB) Klassifikation für AML basierte auf zytomorphologischen Merkmalen und wurde durch die WHO-Klassifikationen 2001/2008 und 2016 ersetzt.
Das European LeukemiaNet (ELN) definiert 3 Risikogruppen nach genetischen Anomalien (Döhner et al. 2017).
Bestimmte AML-Untergruppen wie die akute promyelozytäre Leukämie (APL) (PML-RARA, t) profitieren von einer gezielten Behandlung und haben eine hervorragende Prognose.
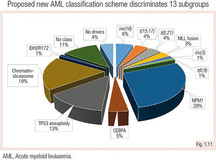
Ein neues Klassifikationsschema wurde vorgeschlagen, das Karyotyp und somatische Mutationen einschließt und 13 AML-Untergruppen definiert (Papaemmanuil et al. 2016).
Spezifische Chromosomenaberrationen wie t(8;21), inv(16), t(15;17) sind krankheitsdefinierend, unabhängig von der quantifizierten Blastenzahl.
In Zukunft könnte sich die Diagnose der AML allein auf genetische Befunde stützen.
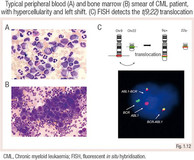
CML ist gekennzeichnet durch eine Leukozytose mit myeloischen Vorläuferzellen im peripheren Blut, die als „Linksverschiebung“ bezeichnet wird. Die Krankheit wird durch das Philadelphia-Chromosom t(9;22) ausgelöst, das das konstitutiv aktive Fusionsprotein BCR-ABL1 produziert.
CML wird je nach Anzahl der Blastenzellen in die chronische Phase, die akzelerierte Phase und die Blastenphase eingeteilt.
Die Therapieüberwachung erfolgt durch hochempfindliche Echtzeit-PCR zum Nachweis von BCR-ABL1.
Fragen zur Revision
- Was ist die Grundlage für die AML-Klassifikation der WHO 2016?
- Welche Aberrationen und Mutationen sind diagnostisch für AML, ohne dass ≥20% Blasten erforderlich sind?
- Was ist die genetische Grundlage der CML?