JOHDANTO
Pitkän QT:n oireyhtymälle (LQTS) on ominaista vakavasti muuttunut kammioperäinen repolarisaatio, joka johtaa QT-intervallin pidentymiseen EKG:ssä. Tila altistaa potilaat pahanlaatuisille kammioperäisille rytmihäiriöille (torsade de pointes) ja äkkikuolemalle. Pitkän QT-ajan oireyhtymän kliinisen ja elektrokardiografisen kuvauksen tekivät vuonna 1957 Anton Jervell ja Fred Lange Nielsen1 , jotka julkaisivat tutkimuksensa perheestä, jonka vanhemmat eivät olleet sukua keskenään ja jossa oli kuusi lasta. Neljällä lapsella oli synnynnäinen kuurous ja synkooppisia jaksoja, ja kolmella lapsella ilmeni äkkikuolema. Näiden potilaiden EKG-tutkimus osoitti epätavallisen pitkän QT-välin. Molemmat vanhemmat olivat oireettomia, EKG oli normaali eikä heillä ollut kuulo-ongelmia. Vuonna 1964 Romano ja Ward raportoivat itsenäisesti sydänoireyhtymästä, jolle oli ominaista toistuvat synkopeet, perheessä esiintynyt äkkikuolema ja QT-intervallin pidentyminen ilman hermokuuroutta.2 Myöhemmin tehdyt geneettiset tutkimukset osoittivat, että Jervellin ja Lange Nielsenin kuvailema oireyhtymä, johon liittyy synnynnäinen hermokuurous, vastaa homotsygoottisia mutaatioita, joilla on vakava fenotyyppi ja suuri äkkikuoleman riski. Romano-Wardin oireyhtymänä tunnettu tila vastaa yleensä heterotsygoottisia mutaatioita, potilailla ei esiinny kuulomuutoksia, ja taudin vaikeusaste vaihtelee huomattavasti. Lähes puoli vuosisataa myöhemmin, vuonna 1995,3,4 kuvattiin tärkeimmät LQTS:ään liittyvät geenit, ja tauti tunnustettiin sydämen ionikanavahäiriöksi. Se oli ensimmäinen kuvattu sydänkanavapatia, ja se on ehkä tähän mennessä laajimmin tutkittu rytmihäiriöitä aiheuttava ionikanavahäiriö. Kliininen kuva vaihtelee suuresti: potilas voi olla oireeton, tai hänellä voi esiintyä toistuvia pyörtymisiä, kouristuskohtauksia tai äkkikuolema taudin ensimmäisenä ilmenemismuotona. Aluksi LQTS:ää pidettiin harvinaisena oireyhtymänä, ja tosiasiassa taudin vaikea esiintyminen on satunnaista. Siitä huolimatta siihen liittyvien mutaatioiden esiintyvyydeksi on arvioitu 1/3000-5000 tapausta,5 32 %:lla oireettomista kantajista sydämen sykekorjattu QT-väli (QTc) voi olla normaalin rajoissa, tauti periytyy 50 %:lle heidän jälkeläisistään, he ovat alttiimpia sairastumaan rytmihäiriöihin verrattuna yleiseen väestöön ja jopa 20 %:lla voi tulla oireita.6
Pitkän QT:n oireyhtymässä on havaittavissa suurta perinnöllistä heterogeenisuutta. Tässä tilassa on kuvattu yli 500 mutaatiota, jotka jakautuvat 10 geeniin: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 ja SCN4B. Vaikka tällä alalla on tapahtunut edistystä, geneettistä diagnoosia ei pystytä asettamaan 25-30 prosentille potilaista.7,8 Sairauden esiintymistapa on pääasiassa monogeeninen6 ; polygeenisten tai yhdistelmätapausten fenotyyppi on yleensä vaikeampi. Penetranssi, eli potilaat, joilla on mutaatio ja joilla ilmenee fenotyyppi, vaihtelee 25 prosentista 90 prosenttiin.9 Harvemmin taudin ekspressiivisyydessä voi olla vaihtelua, jolloin samasta mutaatiosta aiheutuu useita fenotyyppejä. Viimeisten 11 vuoden aikana kehitetyt molekyyligeneettiset tutkimukset ovat tuottaneet tärkeitä genotyypin ja fenotyypin välisiä korrelaatioita, jotka ovat auttaneet ohjaamaan hoitomenetelmiä. Lisäksi on tehty mielenkiintoisia havaintoja yksilöllisestä alttiudesta sairastua rytmihäiriöihin tutkimuksissa, joissa on tutkittu tässä väestössä usein esiintyviä ei-synonyymisiä polymorfismeja, mikä on herättänyt suurta kiinnostusta erityisesti farmakogenomiikan alalla.
PITKÄN QT-PITKÄN QT-SYNDROOMIN LUOKITTELU
Yleisiä käsitteitä
Aiemmin käytetty LQTS-luokitus perustui taudin homotsygoottiseen tai heterotsygoottiseen esitystapaan, jolloin syntyy Jervell-Lange-Nielsenin oireyhtymä (johon liittyy kuuroutuminen) ja Romano-Wardin oireyhtymä (jossa ei ole kuuroutumista). Nykyisessä luokittelussa korostetaan geneettisiä löydöksiä, kuten taulukosta 1 käy ilmi. Kolme tärkeintä tautiin liittyvää geeniä kuvattiin vuosina 1995-1996. Nämä geenit, jotka koodaavat kaliumkanavien IKs ja IKr sekä natriumkanavan Nav1.5 huokosia muodostavia yksiköitä, aiheuttavat lähes 65 prosenttia tapauksista. Vaikka myöhempinä vuosina luetteloon on lisätty seitsemän muuta geeniä, niiden osuus tapauksista on vain 5 %.
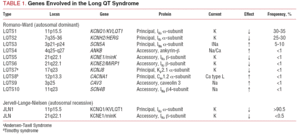
Ionikanavat ovat transmembraaniproteiineja, jotka kuljettavat ioneja solukalvon läpi. LQTS:ään liittyvät kanavat ovat selektiivisiä tai erikoistuneet kuljettamaan yhtä ionia, ja ne ovat jännitteestä riippuvaisia, eli niiden aktivoituminen tapahtuu tietyssä solunsisäisessä jännitteessä, joka vaihtelee kanavan alatyypin mukaan. Kardiomyosyytissä tapahtuvat sähköiset ja supistumisilmiöt ovat näiden rakenteiden ohjaamia. Ionikanavat muodostavat makromolekyylikomplekseja, jotka koostuvat kanavahuokosen muodostavasta pääyksiköstä ja sitä säätelevistä apuproteiineista (kuva 1). LQTS:ssä havaittu kanavan toimintahäiriö voi esiintyä näissä kahdessa kohdassa: pääproteiinissa tai säätelevissä proteiineissa (taulukko 1). Huokosta muodostavan yksikön, niin sanotun alfa-yksikön, osallistuminen synnyttää kolme yleisintä LQTS:n alatyyppiä: LQTS1 (vaikuttaa IKs-kaliumkanavaan), LQTS2 (vaikuttaa IKr-kaliumkanavaan) ja LQTS3 (vaikuttaa natriumkanavaan). Koska nämä ovat yleisimmät alatyypit, ne ovat kliinisesti ja geneettisesti parhaiten karakterisoituja. Fenotyypin ja genotyypin väliset korrelaatiot näissä kolmessa päämuodossa on kuvattu kuvassa 2. Tällä hetkellä Jervell-Lange-Nielsenin oireyhtymä vastaa LQTS 1 ja 5 -muotoja. Näille potilaille on tyypillistä synnynnäinen kuurous ja IKs-virtaan vaikuttavat homotsygoottiset tai heterotsygoottiset mutaatiot. Romano-Wardin oireyhtymään kuuluvat LQTS 1-10 -lajikkeet, eikä siihen liity kuuroutta.
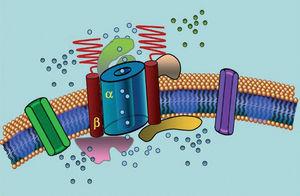
Kuva 1. Makromolekyylikompleksin kaavamainen esitys. Ionikanavat ovat transmembraaniproteiineja (α), joita säätelevät eri proteiinit; yksi niistä on ns. β-alayksikkö.
Pitkän QT:n oireyhtymä tyyppi 1 (LQTS1)
Potilailla, joilla on LQTS1, esiintyy kammioperäisiä rytmihäiriöepisodeja yleensä liikunnan tai sympaattisen ärsykkeen yhteydessä (68 %).10 Uinti on kuvattu urheilulajina, joka laukaisee rytmihäiriöitä LQTS1:ssä.11 Penetranssi on lähes 62 % tässä alatyypissä. Näillä potilailla T-aallon pohja on usein laaja ja kesto hyvin pitkä12,13 (kuva 2). Se on yleisin alatyyppi, ja se selittää 30-35 prosenttia tapauksista. Vaikuttava geeni, KvLQT1 (tai KCNQ1), sijaitsee kromosomissa 11 (11p15.5) ja koodaa IKs-kaliumkanavan α-alayksikköä. Toimintapotentiaali pitkittyy, koska lähtevä K+ -virta vähenee toimintapotentiaalin vaiheessa 3.
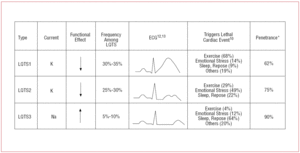
Kuvio 2. Toimintapotentiaali. Genotyypin ja fenotyypin välinen korrelaatio yleisimmissä pitkissä QT-oireyhtymissä. *Viittaa tapauksiin, joissa on mutaatio ja joissa ilmenee fenotyyppi.
Pitkän QT:n oireyhtymä tyyppi 2 (LQTS2)
Potilailla, joilla on LQTS2, esiintyy kammioperäistä rytmihäiriötä yleensä emotionaalisen stressin (49 %) tai äkillisten ääniärsykkeiden (esim. herätyskellon) yhteydessä ja harvemmin unen (22 %) tai liikunnan (29 %) aikana.10 Erityisen alttiita sille ovat synnytyksen jälkihoitoaikana elävät naiset.14 Arvioitu penetraatio on 79 %, joten jopa 20 %:lla tapauksista voi olla diagnosoimaton EKG. LQTS2:n T-aalto on yleensä matalaamplitudinen ja kaksijakoinen, ja siinä on lovia12,13 (kuva 2). Vaikuttava geeni on KCNH2 tai HERG, joka sijaitsee kromosomissa 7 (7q35-36) ja joka koodaa IKr-kaliumkanavan α-alayksikköä, ja sen osuus on 25-30 % tapauksista. Tämän kanavan toimintahäiriö vähentää lähtevää K+ -virtaa toimintapotentiaalin vaiheessa 3 ja pidentää sen kestoa.
Pitkä QT-oireyhtymä tyyppi 3 (LQTS3)
Potilailla, joilla on LQTS3-oireyhtymä, on suurempi riski saada pahanlaatuisia rytmihäiriöitä levossa (unessa) tai bradykardiapotilailla.15 SCN5A-geenin mutaatioiden penetranssisuus on lähes 90 %. LQTS3:n EKG:ssä näkyy yleensä viivästynyt, teräväkärkinen T-aalto, ja siinä voidaan selvästi havaita ST-segmentin pidentyminen12,13 (kuva 2). Näillä potilailla on yleensä vähemmän oireita kuin LQTS1- tai LQTS2-potilailla, mutta tapahtumat ovat tyypillisesti tappavampia.
LQTS3:ssa kyseinen geeni on SCN5A, joka koodaa Nav1.5-natriumkanavan α-alayksikköä (kuva 1), ja se sijaitsee kromosomissa 3 (3p21-24); se on taudin aiheuttaja 5-10 prosentissa tapauksista. Kanavan viallinen inaktivaatio mahdollistaa Na+:n jatkuvan syötön toimintapotentiaalin vaiheessa 2, mikä pidentää sen kestoa.
Pitkän QT:n oireyhtymä tyyppi 4 (LQTS4)
Tyyppi 4 on harvinainen LQTS:n muunnos, jonka osuus on lähes 1 % tapauksista. Se on epätyypillinen muoto, joka tuottaa laajan kirjon rytmihäiriöitä, mukaan lukien katekolaminerginen polymorfinen kammiotakykardia, eteisvärinä, kammionsisäiset johtumismuutokset, sinussolmukkeen toimintahäiriö ja bradykardia6-18; lisäksi QTc voi olla monilla potilailla normaalin rajoissa. Vaikuttava geeni on ANKB, joka sijaitsee kromosomissa 4 (4q25-27) ja joka koodaa ankyrin-β:n synteesiä, rakenteellisen proteiinin, joka yhdistää kardiomyosyyttien kalvoproteiinit sytoskelettiproteiineihin. Näitä proteiineja ovat Na/K-ATPaasi-pumppu, Na/Ca-vaihtaja ja inositolitrifosfaattireseptori (InsP3R). Mutaatiot, jotka aiheuttavat ankyrin-β:n toiminnan menetyksen, johtavat solunsisäisen kalsiumkonsentraation nousuun ja muutoksiin N/K-ATPaasin ja Na/Ca-vaihtimen ilmentymisessä. Kohonnut kalsiumkonsentraatio aiheuttaa varhaisia ja viivästyneitä jälkidepolarisaatioita. Ankyrin-β-geenin mutaatioissa havaitut kammioperäiset rytmihäiriöt johtuvat siis spontaaneista depolarisaatioista, jotka ovat yleensä vaste katekolaminergiselle stimulaatiolle.
Pitkä QT-oireyhtymä tyyppi 5 (LQTS5)
Tyyppi 5 saa alkunsa kromosomissa 21 (21q22.1p22) sijaitsevan KCNE1-geenin sekvenssin muutoksista.19 KCNE1 koodaa IKs-kanavan β-alayksikön, joka tunnetaan myös minK-alayksikkönä, synteesiä, ja se säätelee IKs-kanavaa. Tämän tyypin osuus tapauksista on alle 1 %.
Pitkän QT:n oireyhtymä tyyppi 6 (LQTS6)
Tyypissä 6 vaikuttava geeni on KCNE2, joka sijaitsee kromosomissa 21 (21q22.1.1).20 Tämä geeni koodaa kaliumkanavan β-alayksikön synteesiä, joka tunnetaan myös nimellä MiRP1-alayksikkö, ja se säätelee IKr-kanavaa. Alle 1 % tapauksista on tyypin 6 tapauksia.
Pitkän QT:n oireyhtymä tyyppi 7 tai Andersen-Tawilin oireyhtymä (LQTS7)
Tohtori Andersen21 kuvasi ensimmäisen kerran tämän oireyhtymän dysmorfiset löydökset ja elektrokardiografiset muutokset vuonna 1971, ja tohtori Tawil käsitteli niitä uudelleen vuonna 199422 , mutta geneettinen/molekulaarinen kuvaus raportoitiin vasta vuonna 2001.23 Nykyisin Andersen-Tawilin oireyhtymänä (ATS) tunnettu tila on autosomaalinen dominoiva muutos, jolle on ominaista jaksottainen halvaus, luuston epänormaali kehitys, kammioperäinen rytmihäiriö, johon liittyy usein kammioekstrasystoleja, ja erityinen alttius kammiovärinän kehittymiselle, erityisesti naisilla. ATS:ssä kuvattuihin muutoksiin kuuluvat kammioperäiset ekstrasystoliat (41 %), ei-kestävä polymorfinen kammiotakykardia (23 %), kaksisuuntainen kammiotakykardia (68 %) ja torsade de pointes (3 %).24 Joihinkin havaittuihin dysmorfisiin piirteisiin kuuluvat lyhytkasvuisuus, skolioosi, kliinodaktyylisyys, hypertelorismi, korvien matala asettuminen, mikrognatia ja leveä otsa. Taudin ilmeneminen vaihtelee, mikä vaikeuttaa varhaista diagnoosia.23,25 Mutaatiot kromosomissa 17 (17q23) sijaitsevassa KCNJ2-geenissä, joka koodaa oikaisevan kaliumkanava Kir 2.1:n synteesiä, aiheuttavat 70 prosenttia tapauksista. Tämä kanava osallistuu toimintapotentiaalin 4. vaiheeseen. Useat kirjoittajat kyseenalaistavat tämän geenin sisällyttämisen LQTS:n kausaaliryhmään, koska QTc-väli on tässä oireyhtymässä vain hieman pidentynyt tai jopa normaali, mutta U-aalto on yleensä näkyvä, mikä on johtanut QT-välin yliarvioimiseen. Lukija huomaa, että joidenkin kirjoittajien mukaan KCNJ2-mutaatiot synnyttävät ATS1:n eikä LQTS7:ää.24
Pitkä QT-oireyhtymä tyyppi 8 (LQTS8)
Tyyppi 8 johtuu mutaatioista kromosomissa 12 (12p13.3) sijaitsevassa CACNA1-geenissä, joka koodaa L-tyypin kalsiumkanava Cav1:tä.2. Se aiheuttaa Timothyn oireyhtymän,26 tilan, jolle on ominaista sydämen epämuodostumat, ajoittainen immunologinen puutos, hypoglykemia, kognitiiviset muutokset, mukaan lukien autismi, interdigitaalinen fuusio ja pidentynyt QT-aika, joka johtaa sydämen rytmihäiriöihin ja äkkikuolemaan.27 Alle 0,5 % tapauksista on tyypin 8 tapauksia.
Pitkä QT-oireyhtymä tyyppi 9 (LQTS9)
Tämä LQTS:n muunnos kehittyy mutaatioista kromosomissa 3 (3p25) sijaitsevassa CAV3-geenissä, joka koodaa kaveoliini 3:n synteesiä. Kaveoliini on plasmakalvon invaginaatio, joka osallistuu endosytoosiin, lipidihomeostaasiin ja signaalinsiirtoon. Tärkeä osa tätä rakennetta on kaveoliini, jolla on kolme tunnettua alatyyppiä; alatyyppi 3 on spesifinen luuranko- ja sydänlihaksille. Jotkin ionikanavat sijaitsevat kaveoliinissa, mukaan lukien natriumkanava Nav1.5:n sydämen isomuoto. Tässä proteiinissa on hiljattain kuvattu useita mutaatioita. Ne muuttavat natriumkanava Nav1.5:n biofysikaalisia ominaisuuksia in vitro, jolloin syntyy samanlainen fenotyyppi kuin LQTS3:ssa havaittu.28 Alle 1 % tapauksista johtuu tästä syystä.
Pitkä QT-oireyhtymä, tyyppi 10 (LQTS10)
Tyyppi 10 kuvattiin erittäin vaikeassa tapauksessa, jossa QTc oli >600 ms, sikiöaikainen bradykardia ja eteis-kammiosolmukesalpauksen (AV-blokki) 2:1. Se johtuu mutaatioista SCN4B-geenissä, joka sijaitsee kromosomissa 11 (11q23) ja joka koodaa natriumkanavan β4-alayksikköä. On kuvattu neljä eri β-alayksikön alatyyppiä, jotka ovat vuorovaikutuksessa ja säätelevät eri natriumkanavan isoformeja; kuitenkin vain alatyyppi 4 on tähän mennessä yhdistetty rytmihäiriöiden syntyyn29. Tämän geenin mutaatioiden esiintyvyyttä ei ole tutkittu, mutta sen arvioidaan olevan
Jervell-Lange-Nielsenin muunnoksen mutaatiot
Tämä LQTS:n vakava muoto aiheutuu homotsygoottisista30 tai yhdistelmäheterotsygoottisista mutaatioista KCNQ1- ja/tai KCNE1-geeneissä, jotka koodaavat IKs-virtausta; ts. kyseessä on siis erittäin vakava muunnos LQTS1- tai LQTS5-muodon muunnoksesta. Tähän tilaan liittyy tyypillisesti synnynnäinen kuurous. Potilailla on yleensä QTc>500 ms ja toistuvia pyörtymisiä, ja heillä on suuri äkkikuoleman riski. Tätä muunnosta sairastavien potilaiden vanhemmat ovat yleensä heterotsygoottisia, ja heidän tautinsa on lievempi tai heillä ei ole oireita.31
PITKÄN QT-PITUUDEN SYNDROOMIN DIAGNOSOINTI
Schwartzin pistemäärä
Schwartz ym.32 julkaisivat vuonna 1985 LQTS:n diagnosointikriteerit, joita muokattiin vuonna 1993, ja ne sisältävät tärkeät ohjeet mahdollisten tapausten alustavaa arviointia varten. Tässä järjestelmässä käytetään pistemäärää 1-9, joka perustuu sukuhistoriaan sekä kliinisiin ja elektrokardiografisiin löydöksiin. Taudin todennäköisyys on pieni, kun pistemäärä on ≥1, keskinkertainen, kun pistemäärä on 2-3, ja suuri, kun pistemäärä on ≥4 (taulukko 2).

Pitkän QT:n oireyhtymän synnytyksen aikainen diagnostiikka
Sikiöaikainen bradykardia voi olla yksi ensimmäisistä kliinisistä ilmenemismuodoista, jotka osoittavat, että potilaalla on pitkää QT-hetkielimistöliikkeitä. Retrospektiiviset sarjat ovat osoittaneet, että jopa 70 %:lla lapsuudessa diagnosoidusta LQTS:stä kärsivistä potilaista on ollut bradykardiaa, johon on yleensä liittynyt sikiön hydropsia.33 Sikiön sydämen repolarisaation arviointi viikoilla 14-39 on hyödyllistä LQTS:n varhaisdiagnostiikassa.34
LQTS:n gonadaaliseen mosaiikismiin on liitetty toistuvia sikiön menetyksiä raskauden kolmannella kolmanneksella.35 Lisäksi saostavat tekijät ja synkopeen konteksti voivat osoittaa LQTS:n alatyypin. Epäillyn tapauksen ensiarvioinnissa on suljettava pois sellaisten lääkkeiden käyttö, jotka voivat pidentää QT-väliä.
QT-väli: Mikä on normaalia?
QT-väli tulisi mitata mieluiten johtimista II tai V5,37 joissa sillä on todistetusti suurempi ennustearvo.38 Tämä väli ilmaisee kammion repolarisaation keston ja se mitataan Q-aallon alusta T-aallon loppuun. Perinteisesti käytetään Bazettin39 ehdottamaa kaavaa intervallin keston korjaamiseksi sykkeen mukaan (QTc=QT/√RR, sekunteina ilmaistuna). Vaikka QT-välin mittaaminen vaikuttaa yksinkertaiselta, Viskinin ja muiden tekemässä monikeskustutkimuksessa40 alle 40 prosenttia muista lääkäreistä kuin kardiologeista, alle 50 prosenttia kardiologeista ja yli 80 prosenttia rytmihäiriötautien erikoislääkäreistä osasi mitata QT-välin oikein. On suositeltavaa, että lääkärit suorittavat manuaalisen mittauksen eivätkä luota automaattisiin mittauksiin, jotka voivat olla hyödyllisiä muiden intervallien osalta, mutta ovat epätarkkoja QT-väliä laskettaessa. QT-väli on dynaaminen intervalli, ja sen normaalirajat riippuvat useista tekijöistä. Vaikka miehillä 440 ms:n ja naisilla 460 ms:n QTc-väliä pidetään epänormaalina, mutaatioiden kantajia ja terveitä henkilöitä voi löytyä tältä alueelta (kuva 3). Vincent ja muut41 osoittivat LQTS1-perheissä, että yhdessäkään positiivisen genotyypin omaavassa tapauksessa QTc-intervalli ei ollut 470 ms. Monnig ym.38 osoittivat äskettäin, että QTc>440 ms riittää havaitsemaan potilaat, joilla on LQTS:ään liittyviä mutaatioita, QTc>470 ms on käyttökelpoinen tunnistamaan potilaat, joilla on riski oireiden kehittymiselle, ja QTc>500 ms löytyy oireilevilta potilailta, jotka ovat hoidon piirissä.
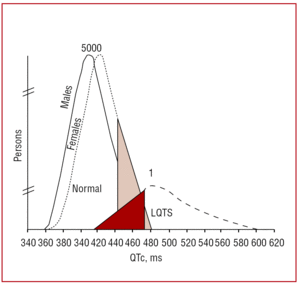
Kuvio 3. LQTS:n mutaatiot. Malli, joka osoittaa sykekorjatun QT-välin (QTc) jakautumisen potilailla, joilla on mutaatioita KVLQT1:ssä, HERG:ssä tai SCN5A:ssa, ja heidän perheenjäsenillään, joilla ei ole mutaatioita. Vasemmalla oleva käyrä kuvaa sairastumattomien perheenjäsenten jakaumaa ja oikealla oleva käyrä sairastuneiden perheenjäsenten jakaumaa.
Muut pitkän QT:n oireyhtymään liittyvät sähkökardiografiset muutokset
Potilailla, joilla on LQTS-oireyhtymä, voi esiintyä useita T-aaltojen muutoksia: muun muassa polariteetin vuorottelua, amplitudin vaihteluita, loveutumista ja bifaasista ilmenemismuotoa.42 T-aallon vuorottelu (kuva 4A) määritellään sinusrytmin T-aallon amplitudin, morfologian ja polariteetin sykekohtaiseksi vaihteluksi ilman QRS-kompleksin vaihtelua. Se on sähköisen epävakauden indikaattori,43 joka heijastaa kammion repolarisaation alueellista hajontaa, ja se edeltää toisinaan kammiovärinää.44
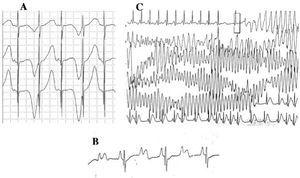
Kuva 4. Kammiovärinä. Elektrokardiografiset muutokset pitkän QT:n oireyhtymässä. A: T-aallon sähköinen vaihtelu. B: eteis-kammiokatkos 2:1. C: itsestään rajoittuva torsade de pointes.
LQTS-potilailla voi edetä sinussolmukkeen toimintahäiriön, bradykardian ja/tai taukojen merkkejä.45 LQTS1- ja LQTS3-alatyypeillä, erityisesti jälkimmäisellä, esiintyy usein sinusbradykardiaa46 , kun taas LQTS4:ään on liitetty sinussolmukkeen toimintahäiriöitä.18
Vuosikymmenestä 1970-1980 lähtien on havaittu AV-johtumishäiriöiden ja LQTS:n47 samanaikaista esiintymistä (kuva 4B). Kaksi yhteen – AV-blokki on harvinainen ilmenemismuoto, jolla on huono ennuste ja joka voi esiintyä sikiövaiheesta lähtien pysyvän bradykardian muodossa. Tämän poikkeavuuden esiintyvyydeksi on raportoitu 4-5 %48 , ja siihen liittyy korkea kuolleisuus beetasalpaajilla ja/tai sydämentahdistimilla annetusta hoidosta huolimatta.49,50 Tämä ilmiö voidaan selittää toimintapotentiaalin pitkällä kestolla. Kun kammioiden refraktorinen jakso pitenee, seuraava sinusaktivaation impulssi estyy, koska se saavuttaa kammiot, kun ne ovat vielä refraktorisessa jaksossa. Tämä muutos näyttää tapahtuvan yksinomaan LQTS:ssä, koska kammioiden refraktorinen aika on pidempi kuin AV-johtumisjärjestelmän.51 QRS-kompleksin kaltevuus on yleensä jyrkkä, ja blokki on paikallistettu infraHis-alueelle,46,51,52 mutta blokin paikka voi riippua genotyypistä. Tähän mennessä neljä geeniä on yhdistetty LQTS:n 2:1-blokkiin: HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 ja SCN4B (LQTS10).55
LQTS:lle ominainen kammioperäinen rytmihäiriö tunnetaan nimellä ”torsade de pointes” eli kammioperäinen rytmihäiriö (kuva 4C). Se ilmenee, kun QT-väli on pidentynyt etiologiasta riippumatta. Se on reentryn aiheuttama polymorfinen kammiotakykardia, jolle on sähkökardiografisesti ominaista QRS-akselin jatkuva kiertyminen kuvitteellisen viivan ympäri. Sitä edeltää yleensä tauko, jota seuraa ekstrasystolia (lyhyt-pitkä-lyhyt RR-väli), kuten kuvassa on esitetty.56-58 Se voi kulminoitua kammiovärinään ja äkkikuolemaan. Jos näin ei tapahdu, potilas voi kokea vain pyörtymisen, ja jos episodi on lyhyt, se voi jäädä huomaamatta.
Holter
Holter-tutkimus antaa täydellisen, dynaamisen arvion QT-välistä. Toisinaan rekisteröidään oireettoman kammioperäisen rytmihäiriön spontaaneja jaksoja sekä sinussolmukkeen toimintahäiriön tai AV-blokin jaksoja.
Rasituskuormituskoe
Potilaat, joilla on LQTS, eivät kykene saavuttamaan iän mukaan laskettua odotettavissa olevaa maksimisykettä. Lisäksi rasituksessa QT-väli voi käyttäytyä paradoksaalisesti pikemminkin pitenemällä kuin lyhenemällä.59,60 Sähkökardiografinen kuvio rasituskuormitustestin aikana on erilainen riippuen LQTS:n tyypistä. Sen lisäksi, että LQTS1-potilaat eivät saavuta ikäisekseen laskettua maksimisykettä, heillä on usein myös QT-väli pidentynyt, kun taas LQTS2-potilaat voivat saavuttaa odotetun sykkeen ja QT-väli pidentyy vain lievästi tai ei lainkaan.61,62 Yleensä LQTS3-potilailla on fysiologinen vaste rasitukseen, eli QT-väli lyhenee normaalisti.63 Rasituskokeet voivat olla hyödyllisiä myös hoitovasteen arvioinnissa ja riskin stratifioinnissa oireettomissa tapauksissa tai silloin, kun on epäilyksiä rytmihäiriöön johtavista tapahtumista.
Geneettinen seulonta
LQTS:n geneettiset tutkimukset ovat viime vuosina rajoittuneet tutkimuslaboratorioihin. Näistä ponnisteluista saadut tiedot ovat kuitenkin olleet erittäin hyödyllisiä potilaiden hoidossa, erityisesti korkean riskin tapauksissa. Seulonnan ehkä tärkein sovellus on geneettinen neuvonta, mutta sillä on myös merkittäviä vaikutuksia hoitoon, jota voidaan suunnata vaikutuskanavan mukaan. Tietyn mutaation tarkka sijainti voi antaa lisätietoa riskin kehittymisestä. Potilailla, joilla on mutaatioita KCNQ1:n transmembraanialueella (IKS), on suurempi todennäköisyys sairastua rytmihäiriöihin kuin potilailla, joilla on mutaatioita C-terminaalialueella64 ; sama pätee potilaisiin, joilla on mutaatioita KCNH2:n tai HERG:n huokosalueella65 verrattuna potilaisiin, joilla on mutaatioita N- tai C-terminaalialueella66 .
Alustava seulonta voidaan ehkä rajoittaa KCNQ1-, HERG- ja SCN5A-geeneihin, jotka tarjoavat mahdollisuuden kohdata mutaatioita 65 prosentissa tapauksista. Kun saadut tulokset ovat negatiivisia, seulontaa voidaan laajentaa KCNE1-, KCNE2-, ANKB-, KCNJ2-, CACNA1-, CAV3- ja SCN4B-geeneihin, mikä lisää positiivisten tulosten mahdollisuutta 5-10 %:lla.
Postmortem-geneettinen seulonta
On mielenkiintoista, että LQTS:ään johtavia geenimutaatioita on löydetty lapsilta, jotka ovat kokeneet äkkikuoleman, ja nuorilla aikuisillakin esiintyvistä selittämättömistä äkkikuolemista.
Kuolemanjälkeisissä geneettisissä tutkimuksissa äkkikuolemapotilailla, joiden ruumiinavaus on ollut negatiivinen, on todettu LQTS:ään johtavia mutaatioita vaihtelevalla prosenttimäärällä67-69: lähes 10 % lapsilla ja 35 % nuorilla aikuisilla.70-72 Näiden tulosten perusteella on ehdotettu rutiininomaista EKG-tutkimusta kaikille vastasyntyneille.73,74
Postmortem-geenitutkimuksella, joka kirjallisuudessa tunnetaan myös nimellä ”molekulaarinen ruumiinavaus”, on oikeudellisten seurausten lisäksi merkittäviä vaikutuksia perheisiin, jotka saattavat sairastua tietämättään.
Säätelyyn liittyvät polymorfismit
LQTS-populaatiossa on kuvailtu useita tiheästi esiintyviä polymorfismeja, jotka ovat jakautuneet melkein kaikkiin tähän tilaan liittyviin geeneihin. Vaikka nämä muutokset eivät ilmeisesti ole patogeenisiä, joillakin voi olla seuraavia vaikutuksia75-78:
1. Synnyttää yksilöllisen alttiuden sairastua rytmihäiriöön.
2. Edistää toisen ei-synonyymisen muutoksen patogeenistä vaikutusta.
3. Vähentää toisen ei-synonyymisen muutoksen patogeenistä vaikutusta.
Tällainen on KCNH2:n (HERG) K897T-polymorfismi, jota esiintyy jopa 15 %:lla väestöstä ja joka ei ole ainoastaan yhteydessä alttiuteen tietyille lääkkeille,79 vaan se myös suosii samassa geenissä esiintyvien mutaatioiden patogeenistä vaikutusta.78 Toinen esimerkki on SCN5A-geenin S1103Y-polymorfismi, jota esiintyy pääasiassa mustilla ja jonka esiintyvyys on lähes 13 prosenttia ja joka liittyy lisääntyneeseen lapsuusiän äkkikuoleman riskiin80.
Mielenkiintoista on, että SCN5A-geenin (joka koodaa Nav1.5-natriumkanavan isomuotoa ihmisillä) tuotteessa on kuvattu kaksi vaihtoehtoista prosessointipaikkaa, jotka tuottavat kahdenlaisia natriumkanavia: toisessa on 2016 aminohappoa, jotka sisältävät glutamiinia 1077-asemassa (Q1077), ja toisessa 2015 aminohappoa, joista puuttuu glutamiini (Q1077del). Näiden vaihtoehtoisten käsittelyjen transkriptejä esiintyy samassa ihmissydämessä suhteessa 2:1, ja useilla usein esiintyvillä polymorfismeilla on erilaisia vaikutuksia kanavan toimintaan riippuen siitä, onko kyseessä Q1077- vai Q1077del-konteksti. Tämä osoitettiin alun perin SCN5A:n H558R-polymorfismilla, jota esiintyy jopa 30 prosentilla väestöstä. Kun H558R ekspressoitiin Q1077-kontekstissa, havaittiin ionivirran voimakas väheneminen.81 Samanlainen vaikutus dokumentoitiin S524Y82-polymorfismilla. Nämä havainnot ovat tarjonneet tekijöitä, jotka selittävät taudin vaihtelevaa vaikeusastetta sekä saman mutaation erilaisia fenotyyppejä, joita on havaittu joissakin perheissä.77
Farmakologinen testaus adrenaliinilla
Farmakologinen testaus matala-annoksisella adrenaliinilla on turvallinen ja käyttökelpoinen vaihtoehto, jonka avulla voidaan paljastaa epäillyt LQTS-tapaukset, joilla on raja-arvojen mukainen QTc. Se on erityisen tehokas havaitsemaan oireettomat LQTS1 -muodot, ja sen herkkyys on 92,5 %, spesifisyys 86 %, positiivinen ennustearvo 76 % ja negatiivinen ennustearvo 96 %. Se voi olla hyödyllinen myös LQTS2:n diagnosoinnissa, mutta sen herkkyys ja spesifisyys ovat heikommat. Se ei ole hyödyllinen LQTS3:n tai muiden LQTS-muotojen diagnostiikassa. Normaalioloissa sympaattinen stimulaatio indusoi IKs-kaliumkanavan fosforylaation, joka optimoi sen toiminnan ja johtaa toimintapotentiaalin lyhenemiseen. LQTS-potilailla, erityisesti tyypin 1 potilailla, on havaittu paradoksaalinen vaste matala-annoksisen adrenaliinin (0,025-0,2 µg/kg/min) antoon, joka pidentää QT-väliä yli 30 ms83-86.
QT-VÄLIN PITENEMINEN JA LÄÄKKEIDEN AIHEUTTAMAT TORSADE DE POINTES
Valjon erilaiset eri lääketieteen erikoisaloilla käytettävät lääkkeet voivat aiheuttaa iatrogeenisen QT-intervallien pidentymisen. Joitakin lääkkeitä on poistettu markkinoilta tämän ei-toivotun vaikutuksen vuoksi (esim. mm. astemitsoli ja sisapridi; lisätietoja osoitteessa www.qtdrugs.org).87,88
Muiden kuin rytmihäiriölääkkeiden aiheuttamaa sekundaarista kammioperäistä rytmihäiriötä esiintyy harvemmalla kuin yhdellä altistuneista henkilöistä 10 000-100 000:sta. Kun otetaan huomioon, että kliinisissä tutkimuksissa on mukana 2000-3000 koehenkilöä, tämä ei-toivottu ja kuolemaan johtava haittatapahtuma jää helposti huomaamatta lääkekehityksen kliinisessä vaiheessa.89. Tämä seikka on herättänyt valtavaa kiinnostusta turvallisuuteen liittyviin näkökohtiin uusien lääkkeiden tutkimisessa ja kehittämisessä.
Yksilölliseen alttiuteen liittyviä tekijöitä ovat muun muassa naissukupuoli, hypokalsemia, hypomagnesemia, bradykardia, sydämen vajaatoiminta, postkardioversion jälkeinen sydämen vajaatoiminta, eteisvärinä, vasemman kammion hypertrofia, havaitsematta jäänyt LQTS:n aiheuttama oireyhtymä, altistavat polymorfismit ja altistavien lääkeaineiden korkeat seerumikonsentraatiot.90
Kanava, joka on tyypillisesti vuorovaikutuksessa lääkkeiden kanssa, on molekyylirakenteensa vuoksi KCNH2(HERG)-geenin koodaama IKr. Muissa kaliumkanavissa on 2 proliinijäännöstä kulmautuneena kohti kanavan huokosta, mikä pienentää sen luumenia. Sitä vastoin IKr:stä puuttuvat nämä jäännökset, syntyy suurempi huokosen eteinen ja altistuminen suurille molekyyleille helpottuu. Lisäksi siinä on kaksi aromaattista residuaalia (tyrosiini ja fenyylialaniini), jotka suosivat sitoutumista aromaattisiin molekyyleihin, joita esiintyy useissa kanavaa salpaavissa lääkkeissä.91
Kuten edellä mainittiin, LQTS:n läpäisevyys on epätäydellinen, ja joillakin mutaatioiden oireettomilla kantajilla saattaa ilmetä pahanlaatuista rytmihäiriötä, kun he saavat jonkun näistä lääkkeistä. Lisäksi polymorfismit, joita pidetään väestössä yleisinä, antavat yksilöllisen alttiuden torsade de pointesin kehittymiselle joidenkin lääkkeiden käytön yhteydessä. Tämä koskee R1047L-polymorfismia, joka on toiseksi yleisin KCNH2-geenin polymorfismi ja joka on yhdistetty torsade de pointes -oireyhtymän kehittymiseen dofetilidi-lääkkeen käytön yhteydessä.92 Terveillä henkilöillä on kuvattu ainakin 20 KCNH2-geenin polymorfismia, ja niiden vaikutus yksilölliseen alttiuteen sairastua lääkeaineisiin liittyviin rytmihäiriöihin on vielä määrittelemättä.93 Kammioperäisen rytmihäiriön kehittymiselle altistavia polymorfismeja on havaittu myös natriumkanava Na1.5:ssä. Tällainen on H558R-polymorfismi, jota esiintyy jopa 30 prosentilla väestöstä, tai S1103Y-polymorfismi, jota esiintyy usein mustilla80,81,90,94,95; niiden vaikutusta lääkkeiden aiheuttamaan alttiuteen ei ole tutkittu.
PITKÄ QT-SYNDROOMA JA RASKAUS
Geneettinen neuvonta on tärkeää LQTS:ssä, mutta yleisesti ottaen kantajina olevilla naisilla ei ole vasta-aihetta raskaudelle, vaikka jokainen tapaus on erilainen, ja se on arvioitava yksilöllisesti asianmukaisessa yhteydessä.
On todettu, että pahanlaatuisen kammioperäisen sydämen rytmihäiriön riski pienenee raskauden myötä. Sitä vastoin on raportoitu suuremmasta alttiudesta saada pahanlaatuinen rytmihäiriö synnytyksen jälkeisten yhdeksän ensimmäisen kuukauden aikana, erityisesti potilailla, joilla on LQTS2. Tämä riski pienenee huomattavasti beetasalpaajahoidon myötä.96
RISKIN STRATIFIKAATIO
LQTS:n kehittyminen vaihtelee, ja siihen vaikuttavat QTc-välin kesto, ympäristötekijät, ikä, genotyyppi ja hoitovaste.97,98 Kammioperäiset rytmihäiriöt ovat yleisempiä LQTS1- ja LQTS2-oireyhtymissä, mutta vaikeampia LQTS3-oireyhtymässä.99 Kuten edellä mainittiin, naiset ovat erityisen alttiita pahanlaatuisille rytmihäiriöille synnytyksen jälkeisenä aikana.14
Pitkän QT-ajan oireyhtymää on pidettävä suuririskisenä silloin, kun siihen yhdistetään seuraavat tekijät:
1. Synnynnäinen kuurous (Jervell-Lange-Nielsenin oireyhtymä).
2. Pahanlaatuisesta kammiotakyarytmiasta johtuva toistuva synkopee.
3. Perheessä on esiintynyt äkkikuolemia.
4. Äkillinen kuolema. QTc>500 ms.
5. 2:1 atrioventrikulaarinen blokki.
6. Kammiokatkos. T-aallon sähköinen vuorottelu.
7. LQTS3-genotyyppi.
Priorin ym.97 647 potilaalla tekemä tutkimus osoitti, että todennäköisyys saada vakava tapahtuma (synkopee, sydänpysähdys, äkkikuolema) ennen 40 vuoden ikää on suuri (>50 %), kun QTc on >500 ms LQTS1:llä, LQTS2:lla ja miehillä, joilla on LQTS3. Äskettäin raportoitiin kansainvälisen LQTS-rekisterin analyysi. Äkillisen kuoleman riskiä analysoitiin 2772 nuorella, joilla oli kyseinen sairaus, ja tunnistettiin kolme tekijää, jotka liittyvät suurempaan riskiin tässä väestössä: QTc>530 ms, aiempi synkopee viimeisten 10 vuoden aikana ja sukupuoli. 10-12-vuotiailla pojilla riski oli suurempi kuin tytöillä, mutta 13-20-vuotiailla riski oli vertailukelpoinen.100
HOITO
Oireisilla potilailla, jotka eivät saa hoitoa, on 20 prosentin vuotuinen kuolleisuus ja 50 prosentin kymmenvuotiskuolleisuus ensimmäisen kammioperäisen rytmihäiriötapauksen jälkeen. Vaikka on selvää, että hoito on aloitettava oireiden ilmaantuessa, oireettomien potilaiden hoidossa käytettävästä lähestymistavasta käydään edelleen keskustelua. On dokumentoitu, että sydänpysähdys voi olla taudin ensimmäinen ilmenemismuoto 9 prosentilla potilaista48 , ja että 12 prosentille oireettomista potilaista kehittyy oireita ja he saattavat kokea äkkikuoleman. Aloitushoito beetasalpaajilla olisi aloitettava kaikille LQTS-potilaille. Liikunnan rajoittaminen on suositeltavaa, mutta kliiniset ja elektrokardiografiset riskimerkit ovat hyödyllinen perusta päätöksenteolle. On tärkeää kertoa potilaille riskistä, joka liittyy useiden sellaisten lääkkeiden käyttöön, jotka voivat pidentää QT-väliä ja edistää kammioperäisten rytmihäiriöiden kehittymistä, kuten edellä on mainittu. Sen lisäksi, että geneettinen diagnoosi mahdollistaa tautiin liittyvän asianmukaisen perheneuvonnan, se auttaa arvioimaan ennustetta ja suuntaamaan erityishoitoa.
Beetasalpaajat
Beetasalpaajat ovat LQTS:n ensilinjan hoitoa, ja kaikkien potilaiden olisi saatava niitä alkuhoitona.101 Ne vähentävät sydän- ja verisuonitapahtumien riskiä jopa 64 %100 ja ovat erityisen tehokkaita potilailla, joilla on IKs-kanavamutaatioita (LQTS1),102 joita sympaattinen järjestelmä säätelee suurelta osin. Beetasalpaajat eivät muuta QT-väliä vaan sen sijaan sen hajontaa.103 Vaikka nämä lääkkeet vähentävät tapahtumien esiintyvyyttä,104,105 on osoitettu, että 10 %:lla potilaista, joilla on LQTS1, 23 %:lla potilaista, joilla on LQTS2, ja 32 %:lla potilaista, joilla on LQTS3, esiintyy hoidosta huolimatta kardiovaskulaarisia oireita.106 Erityisesti LQTS3-potilaat eivät näytä saavan merkittäviä hyötyjä; itse asiassa tätä lääkeryhmää olisi käytettävä varoen näillä potilailla, koska kammioperäiset rytmihäiriöepisodit ovat LQTS3:lla yleisempiä, kun sydämen syke on alhainen. Yleisesti ottaen 32 %:lla oireilevista potilaista oireet toistuvat viiden ensimmäisen vuoden aikana ennen beetasalpaajahoidon aloittamista, ja 14 %:lla äkkikuolemakohtauksesta pelastuneista potilaista ilmenee uusi samanlainen tapahtuma viiden vuoden kuluessa, jos he saavat vain tätä hoitoa.107 LQTS:n hoidossa on käytetty useita beetasalpaajia, pääasiassa nadololia (0,5-1 mg/kg/vrk), propranololia (2-4 mg/kg/vrk), metoprololia (0,5-1 mg/kg/vrk) ja atenololia (0,5-1 mg/kg/vrk). Atenololista ei kuitenkaan välttämättä ole hyötyä LQTS:ssä; on ilmoitettu, että ainakin 75 % potilaista, jotka eivät vastanneet beetasalpaajahoitoon, saivat atenololia, joskin tämä havainto voi liittyä suboptimaalisten annosten käyttöön.104 Liikuntakokeet ovat hyödyllisiä sopivan annoksen määrittämiseksi. Maksimisyke ei saisi ylittää 130 lyöntiä/min hoidon aikana.
Natriumkanavan salpaajat
LQTS3:a aiheuttavat natriumkanavan mutaatiot aiheuttavat kanavan viallisen inaktivaation; natriumkanavan salpaajat ovat osoittautuneet hyödyllisiksi näillä potilailla. Flekainidilla tehdyissä tutkimuksissa on dokumentoitu parannuksia syketaajuudessa, T-aaltomuutoksissa ja QT-välissä.108 Myös meksiletiinin on raportoitu parantavan elektrokardiografisia riskimerkkejä.63,109,110 Ranolatsiinilla tehdyissä in vitro -tutkimuksissa on havaittu ihmisillä raportoitujen mutaatioiden haitallisten vaikutusten vähenemistä.111 Vaikka tulokset ovatkin rohkaisevia, on muistettava, että tätä hoitoa arvioivia pitkäaikaistutkimuksia ei ole olemassa eikä raportoituja löydöksiä ole saatu suurista sarjoista. Natriumkanavien salpaajia ei tulisi antaa, jos ei ole vahvistettua geneettistä diagnoosia.
Kaliumin lisäravinteet ja sen saatavuutta lisäävät lääkkeet
Kaliumlisät ja/tai kaliumia säästävät lääkkeet, kuten spironolaktoni, lyhentävät QTc-väliä 24 %:ssa tapauksista.112,113 Kaliumkanavien avautumista suosivat lääkkeet, kuten aprikalim, levcromakalim, nikorandil ja pinasiili, ovat osoittautuneet hyödyllisiksi LQTS:n hoidossa. Alatyypit, joissa niistä on erityistä hyötyä, ovat LQTS1 ja LQTS2.114
Tahdistimet ja defibrillaattorit
Tahdistimen stimulaatiota on käytetty potilailla, joilla on tauko-riippuvainen rytmihäiriö.115,116 Potilaat, joilla on LQTS3, hyötyvät yleensä enemmän tästä hoidosta, koska bradykardian esiintyvyys on tässä ryhmässä suurempi. DDD-tahdistus on tarkoitettu potilaille, joilla on tauko-riippuvainen rytmihäiriö tai korkea-asteinen 2:1 AV-blokki. Alle 70 lyöntiä/min117 ohjelmoidut taajuudet eivät ole hyödyllisiä kammioperäisten rytmihäiriöiden ehkäisemiseksi. On suositeltavaa ohjelmoida anturi nopeaan vasteeseen, koska näillä potilailla syke yleensä kiihtyy epätarkoituksenmukaisesti liikunnan seurauksena. Kaikki toiminnot, jotka edellyttävät taukoja, kuten hystereesi- ja yöaikaiset toiminnot, on kytkettävä pois päältä. PARP:n (postventrikulaarinen eteisen tulenkestoaika) on oltava mahdollisimman lyhyt. Taajuuden säätötoiminnon olisi oltava päällä ekstrasystolisen tauon jälkeisen tauon estämiseksi. On muistettava, että T-aallon ylitunnistaminen ja kaappaushäiriöt voivat myös aiheuttaa taukoja. Implantoitavan kardioverteridefibrillaattorin (ICD) ja beetasalpaajien yhdistetty käyttö vähentää huomattavasti äkkikuoleman esiintyvyyttä.118-120 Näiden toimenpiteiden käyttöaihe on selvä korkean riskin tapauksissa.121 Laitteen ohjelmointi vaihtelee yksittäisen potilaan tarpeiden mukaan, mutta yleisesti ottaen hoidon antamista oireettomissa, itsestään rajoittuvissa tapahtumissa olisi vältettävä; tätä varten suositellaan 15 sekunnin havaitsemisjaksoa. Rytmihäiriöt ovat AID-hoidon komplikaatio. Lähes 15 %:lla potilaista voi esiintyä tämä komplikaatio, joka johtuu suurelta osin ICD-iskun jälkeisestä lisääntyneestä sympaattisesta tonuksesta.118 Tätä ongelmaa voidaan hallita lisäämällä beetasalpaajan annosta. Jos tästä toimenpiteestä ei ole hyötyä, on harkittava sympaattisen ketjun ganglioiden resektiota.
Vasen sympatektomia
Vuonna 1971 esiteltiin sympaattisen gangliektomia hyödyllisenä hoitovaihtoehtona näillä potilailla.122 Vuonna 1991 Schwartz ym.123 julkaisivat ensimmäisen sarjan, jossa oli 85 potilasta, joiden vaste beetasalpaajahoitoon oli huono, joille tehtiin vasen stetektomia rohkaisevin tuloksin: viiden vuoden eloonjäämisprosenttiosuus oli 94 %. Nykyisin tätä hoitovaihtoehtoa tarjotaan suuren riskin potilaille, joilla pyörtyminen jatkuu beetasalpaajahoidosta ja/tai sydämentahdistimen implantoinnista huolimatta, sekä potilaille, jotka saavat usein sähköiskuja implantoidusta defibrillaattorista. Toimenpide koostuu stellate ganglion inferiorisen osan ja sympaattisen ketjun vasemman rintakehän T2-T4 ganglion resektiosta, koska pelkkä vasen stellektomia ei ole osoittautunut riittävän tehokkaaksi. Mikroinvasiivista thorakoskopiaa124,125 on käytetty hyvillä tuloksilla. Suurin tällä menetelmällä hoidettujen potilaiden sarja raportoitiin hiljattain, ja se osoitti, että synkopee-episodien tai äkkikuolemien määrä väheni merkittävästi ja että viiden vuoden elossaoloprosentti oli 95 prosenttia. Potilailla, joilla oli ollut aiempi synkopee, viiden vuoden elossaoloaika oli 97 %, ja 11 %:lla oli mahdollisuus uusintatapahtumaan, joka suurimmalla osalla koostui yhdestä synkopeesta. Myös QT-segmentti lyheni merkittävästi vasemmanpuoleisen sympatektomian jälkeen. Näistä suotuisista tuloksista huolimatta äkkikuoleman ehkäisy ei ole täydellistä, mutta se on vähentynyt 3 prosenttiin. Niillä ICD-potilailla, joille tehtiin leikkaus useiden defibrillaattorisokkien vuoksi, tapahtumien keskimääräinen määrä väheni 25:stä 0:aan, mikä on 95 prosentin vähennys. Hyödyllinen vaikutus vahvistettiin LQTS1:ssä. Hyödyt ovat todennäköisesti pienemmät potilailla, joilla on LQTS2, ja LQTS3:ssa sen tehoa ei ole osoitettu.126
Ablaatio
On raportoitu, että kammioperäisen rytmihäiriön joissakin tapauksissa käynnistävän ekstrasystolian ablaatio voi vähentää kohtausten esiintyvyyttä.127 Pitkäaikaistutkimuksia, joissa olisi ollut riittävä määrä potilaita, ei kuitenkaan ole, jotta tämän tekniikan rutiinikäyttö olisi perusteltua.
Ks. pääkirjoitus sivuilla 675-82
LYHENTEET
AV: atrioventrikulaarinen
AID: automaattinen implantoitavissa oleva defibrillaattori
ECG: elektrokardiogrammi
QTc: sydämen syketaajuudella korjattu QT-aika
ATS: Andersen-Tawilin oireyhtymä
LQTS: pitkä QT-oireyhtymä
Tohtori Medeiros saa taloudellista tukea CONACyT:ltä ja FUNSALUD:lta.