BEVEZETÉS
A hosszú QT-szindrómát (LQTS) súlyosan megváltozott kamrai repolarizáció jellemzi, ami az EKG-n a QT-intervallum megnyúlását eredményezi. Az állapot rosszindulatú kamrai ritmuszavarra (torsade de pointes) és hirtelen halálra hajlamosítja a betegeket. A hosszú QT-szindróma klinikai és elektrokardiográfiai leírását 1957-ben Anton Jervell és Fred Lange Nielsen1 közölte, akik egy 6 gyermekes, nem vérszerinti szülőkből álló családon végzett vizsgálataikat publikálták. A gyermekek közül négynek veleszületett süketsége és szinkópás epizódjai voltak, 3 gyermeknél pedig hirtelen halál következett be. E betegek EKG-vizsgálata szokatlanul hosszú QT-intervallumot mutatott. Mindkét szülő tünetmentes volt, EKG-juk normális volt, és nem mutattak hallásproblémát. 1964-ben Romano és Ward egymástól függetlenül beszámolt egy kardiális szindrómáról, amelyet visszatérő syncope, a családban előforduló hirtelen halál és a QT-intervallum megnyúlása jellemez, neuronális süketség nélkül.2 Később genetikai vizsgálatok kimutatták, hogy a Jervell és Lange Nielsen által leírt szindróma, amely veleszületett neuronális süketséggel jár, homozigóta mutációknak felel meg, súlyos fenotípussal és a hirtelen halál magas kockázatával. A Romano-Ward-szindrómaként ismert állapot általában heterozigóta mutációknak felel meg, a betegek nem mutatnak halláselváltozást, és a betegség súlyossága jelentősen változik. Csaknem fél évszázaddal később, 1995-ben3,4 leírták az LQTS-hez kapcsolódó fő géneket, és a betegséget szívioncsatorna-rendellenességként ismerték el. Ez volt az első leírt kardiális csatornelopátia, és talán a mai napig a legkiterjedtebben vizsgált aritmogén ioncsatorna-rendellenesség. A klinikai kép igen változatos: a beteg lehet tünetmentes, vagy a betegség első megnyilvánulásaként visszatérő syncope, rohamok vagy hirtelen halál jelentkezhet. Kezdetben az LQTS-t ritka szindrómának tartották, és valójában a betegség súlyos megjelenése sporadikus. Ennek ellenére a kapcsolódó mutációk előfordulási gyakoriságát 1/3000-5000 esetre becsülik,5 a tünetmentes hordozók 32%-ánál a szívfrekvenciával korrigált QT-intervallum (QTc) a normális határértékeken belül lehet, a betegség leszármazottaik 50%-ára öröklődik, az általános populációhoz képest hajlamosabbak a ritmuszavar kialakulására, és akár 20%-uk tünetessé válhat.6
A hosszú QT-szindróma nagy genetikai heterogenitást mutat. Több mint 500, 10 génben elosztott mutációt írtak le ebben az állapotban: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 és SCN4B. A területen elért eredmények ellenére a betegek 25-30%-ánál nem lehet genetikai diagnózist felállítani.7,8 A betegség megjelenése elsősorban monogén6; a poligénes vagy összetett változatok általában súlyosabb fenotípussal járnak. A penetráció, azaz a mutációval rendelkező és a fenotípust manifesztáló betegek aránya 25% és 90% között mozog.9 Ritkábban a betegség expresszivitásában is lehetnek eltérések, és ugyanabból a mutációból több fenotípus is kialakulhat. Az elmúlt 11 évben végzett molekuláris genetikai vizsgálatok fontos genotípus-fenotípus összefüggéseket mutattak ki, amelyek segítettek a kezelési megközelítés irányításában. Emellett érdekes megfigyelések születtek a ritmuszavar kialakulására való egyéni fogékonyságról az e populációban gyakori nem szinonim polimorfizmusokat vizsgáló tanulmányok során, ami jelentős érdeklődést váltott ki, különösen a farmakogenomika területén.
A HOSSZÚ QT-SZÍNDRÓMA OSZTÁLYZÁSA
Általános fogalmak
A korábban használt LQTS osztályozás a betegség homozigóta vagy heterozigóta megjelenési formáján alapult, amelyből a Jervell-Lange-Nielsen-szindróma (süketséggel), illetve a Romano-Ward-szindróma (süketség nélkül) következik. A jelenlegi osztályozás a genetikai leletekre helyezi a hangsúlyt, amint azt az 1. táblázat szemlélteti. A betegséggel összefüggésbe hozható 3 fő gént 1995-1996-ban írták le. Ezek a gének, amelyek az IKs és IKr káliumcsatornák pórusképző egységeit, valamint a Nav1.5 nátriumcsatornát kódolják, az esetek közel 65%-át teszik ki. Bár a következő években további hét gént vettek fel a listára, ezek az eseteknek csak 5%-át teszik ki.
Az ioncsatornák olyan transzmembránfehérjék, amelyek ionokat szállítanak a sejtmembránon keresztül. Az LQTS-ben szerepet játszó csatornák szelektívek vagy egy-egy ion szállítására specializálódtak, és feszültségfüggőek, azaz aktivációjuk egy adott intracelluláris feszültségnél következik be, amely a csatorna altípusától függően változik. A szívizomsejtben lejátszódó elektromos és kontraktilis jelenségeket ezek a struktúrák irányítják. Az ioncsatornák makromolekuláris komplexeket alkotnak, amelyek a csatorna pórusát alkotó főegységből és az azt szabályozó segédfehérjékből állnak (1. ábra). Az LQTS-ben megfigyelhető csatornadiszfunkció e két helyen fordulhat elő: a főfehérjénél vagy a szabályozó fehérjéknél (1. táblázat). A pórusképző egység, az alfa nevű egység érintettsége hozza létre az LQTS három leggyakoribb altípusát: LQTS1 (az IKs káliumcsatornát érinti), LQTS2 (az IKr káliumcsatornát érinti) és LQTS3 (a nátriumcsatornát érinti). Mivel ezek a leggyakoribb altípusok, klinikailag és genetikailag is ezek a legjobban jellemzettek. E három fő forma fenotípus-genotípus összefüggéseit a 2. ábra mutatja be. Jelenleg a Jervell-Lange-Nielsen-szindróma az LQTS 1 és 5 fajtáknak felel meg. Jellemzően ezeknél a betegeknél veleszületett süketség és az IKs áramot befolyásoló összetett homozigóta vagy heterozigóta mutációk fordulnak elő. A Romano-Ward-szindróma az LQTS 1-től 10-ig terjedő fajtákat foglalja magában, és nem jár süketséggel.
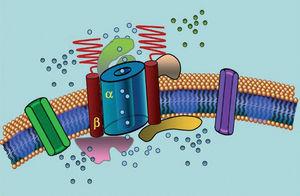
1. ábra. A makromolekuláris komplex sematikus ábrázolása. Az ioncsatornák transzmembrán fehérjék (α), amelyeket különböző fehérjék szabályoznak; az egyik az ún. β alegység.
1. típusú hosszú QT-szindróma (LQTS1)
Az LQTS1-es betegeknél általában sportoláskor vagy szimpatikus inger hatására jelentkeznek kamrai aritmiás epizódok (68%).10 Az úszást az LQTS1-ben ritmuszavart kiváltó sportként írták le.11 A penetráció közel 62%-os ebben az altípusban. A T-hullám ezeknél a betegeknél gyakran széles bázisú és nagyon elhúzódó időtartamú12,13 (2. ábra). Ez a leggyakoribb altípus, és az esetek 30-35%-át magyarázza. Az érintett gén, a KvLQT1 (vagy KCNQ1) a 11. kromoszómán (11p15.5) található, és az IKs káliumcsatorna α-alegységét kódolja. Az akciós potenciál az akciós potenciál 3. fázisában a kimenő K+-áram csökkenése miatt meghosszabbodik.
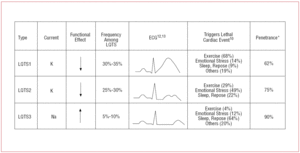
2. ábra. Genotípus-fenotípus korreláció a leggyakoribb hosszú QT-szindrómákban. *A mutációval rendelkező és a fenotípust manifesztáló esetekre vonatkozik.
2. típusú hosszú QT-szindróma (LQTS2)
Az LQTS2-ben szenvedő betegeknél a kamrai aritmia általában érzelmi stresszre (49%) vagy hirtelen hallási ingerre (pl. ébresztőóra), ritkábban alvás (22%) vagy testmozgás (29%) során jelentkezik.10 A szülés utáni időszakban a nők különösen érzékenyek.14 A becsült penetrancia 79%; így az esetek akár 20%-ánál is előfordulhat nem diagnosztikus EKG. Az LQTS2-ben a T-hullám általában alacsony amplitúdójú és kétágú, bevágásokkal12,13 (2. ábra). Az érintett gén a 7. kromoszómán (7q35-36) található KCNH2 vagy HERG, amely az IKr káliumcsatorna α-alegységét kódolja, és az esetek 25-30%-ában fordul elő. E csatorna diszfunkciója csökkenti a kimenő K+ áramot az akciós potenciál 3. fázisában, meghosszabbítva annak időtartamát.
3. típusú hosszú QT-szindróma (LQTS3)
Az LQTS3-ban szenvedő betegeknél nagyobb a kockázata annak, hogy nyugalomban (alvás) vagy bradycardia során malignus aritmiák jelentkeznek.15 Az SCN5A génmutáció penetrációja közel 90%. Az LQTS3-ban az EKG általában késleltetett, hegyes T-hullámot mutat, és egyértelműen megfigyelhető az ST-szakasz megnyúlása12,13 (2. ábra). Ezeknek a betegeknek általában kevesebb tünetük van, mint az LQTS1 vagy LQTS2 betegeknél, de az események jellemzően halálosabbak.
Az LQTS3-ban érintett gén az SCN5A, amely a Nav1.5 nátriumcsatorna α-alegységét kódolja (1. ábra), a 3. kromoszómán (3p21-24) található; az esetek 5-10%-ában ez okozza a betegséget. A csatorna hibás inaktivációja lehetővé teszi a Na+ tartós bevitelét az akciós potenciál 2. fázisában, meghosszabbítva annak időtartamát.
A 4. típusú hosszú QT-szindróma (LQTS4)
A 4. típus az LQTS ritka változata, az esetek közel 1%-át teszi ki. Ez egy atipikus forma, amely az aritmiák széles spektrumát produkálja, beleértve a katekolaminerg polimorf kamrai tachikardiát, pitvarfibrillációt, intraventrikuláris vezetési elváltozásokat, sinuscsomó diszfunkciót és bradycardiát6-18; emellett a QTc sok betegnél a normális határokon belül lehet. Az érintett gén az ANKB, amely a 4. kromoszómán (4q25-27) található, és az ankyrin-β szintézisét kódolja, amely egy olyan strukturális fehérje, amely a kardiomiocita membránfehérjéket a citoszkeletális fehérjékhez kapcsolja. Ezek a fehérjék a Na/K ATPáz pumpa, a Na/Ca cserélő és az inozitol-trifoszfát receptor (InsP3R). Az ankyrin-β funkció elvesztését okozó mutációk az intracelluláris kalciumkoncentráció növekedéséhez és a N/K ATPáz és a Na/Ca cserélő expressziójának megváltozásához vezetnek. A megemelkedett kalciumkoncentráció korai és késleltetett utóddepolarizációt eredményez. Így az ankyrin-β génmutációkban megfigyelt kamrai ritmuszavarok spontán depolarizációkra vezethetők vissza, általában katecholaminerg stimulációra adott válaszként.
Az 5-ös típusú hosszú QT-szindróma (LQTS5)
Az 5-ös típus a 21. kromoszómán (21q22.1p22.)19 található KCNE1 gén szekvenciájának megváltozásával jön létre. A KCNE1 az IKs csatorna β-alegységének, más néven minK-alegységének szintézisét kódolja, amely az IKs csatornát szabályozza. Ez a típus az esetek kevesebb mint 1%-át teszi ki.
6. típusú hosszú QT-szindróma (LQTS6)
A 6. típusban az érintett gén a KCNE2, amely a 21. kromoszómán (21q22.1) található.20 Ez a gén a káliumcsatorna β-alegységét, más néven a MiRP1 alegységet kódolja, és az IKr csatornát szabályozza. Az esetek kevesebb mint 1%-a 6-os típusú.
7-es típusú hosszú QT-szindróma vagy Andersen-Tawil-szindróma (LQTS7)
Az ebben a szindrómában megfigyelhető diszmorfikus leleteket és elektrokardiográfiás elváltozásokat először 1971-ben írta le Dr. Andersen21 , majd 1994-ben Dr. Tawil22 újravizsgálta, de a genetikai/molekuláris leírás csak 2001-ben jelent meg.23 A ma Andersen-Tawil-szindróma (ATS) néven ismert állapot egy autoszomális domináns elváltozás, amelyet periodikus bénulás, rendellenes csontozatfejlődés, gyakori kamrai extraszisztolákkal járó kamrai aritmia és a kamrafibrilláció kialakulására való különös hajlam jellemez, különösen nőknél. Az ATS-ben leírt elváltozások közé tartoznak a kamrai extraszisztolák (41%), a nem fenntartott polymorf kamrai tachycardia (23%), a kétirányú kamrai tachycardia (68%) és a torsade de pointes (3%).24 A megfigyelt diszmorf jellegzetességek közé tartozik a rövid termet, a skoliózis, a klinodaktíliás jelleg, a hipertelorizmus, a fülek alacsony beültetése, a mikrognathia és a széles homlok. A betegség kifejeződése változó, ami megnehezíti a korai diagnózist.23,25 A 17. kromoszómán (17q23) található KCNJ2 gén mutációja, amely a Kir 2.1 egyenirányító káliumcsatorna szintézisét kódolja, az esetek 70%-át teszi ki. Ez a csatorna az akciós potenciál 4. fázisában vesz részt. Több szerző megkérdőjelezi ennek a génnek az LQTS okozati csoportjába való felvételét, mivel a QTc-intervallum ebben a szindrómában csak enyhén megnyúlt vagy akár normális, de az U-hullám általában kiemelkedő, ami a QT-intervallum túlbecsléséhez vezetett. Az olvasó azt találja, hogy egyes szerzők szerint a KCNJ2 mutációi az ATS1-et és nem az LQTS7-et hozzák létre.24
A 8-as típusú hosszú QT-szindróma (LQTS8)
A 8-as típus a 12. kromoszómán (12p13.3) található CACNA1 gén mutációiból ered, amely a Cav1 L-típusú kalciumcsatornát kódolja.2. Ez okozza a Timothy-szindrómát,26 egy olyan állapotot, amelyet szívfejlődési rendellenességek, időszakos immunológiai hiányosság, hipoglikémia, kognitív elváltozások, beleértve az autizmust, interdigitális fúzió és meghosszabbodott QT-idő jellemez, ami szívritmuszavarhoz és hirtelen halálhoz vezet.27 Az esetek kevesebb mint 0,5%-a 8-as típusú.
9-es típusú hosszú QT-szindróma (LQTS9)
Az LQTS ezen változata a 3. kromoszómán (3p25) található CAV3 gén mutációiból alakul ki, amely a kaveolin 3 szintézisét kódolja. A caveolin a plazmamembrán invaginációja, amely szerepet játszik az endocitózisban, a lipid homeosztázisban és a jelátvitelben. E struktúra fontos összetevője a kaveolin, amelynek 3 altípusa ismert; a 3. altípus a váz- és szívizomzatra specifikus. Néhány ioncsatorna a kaveolinnal együtt helyezkedik el, köztük a Nav1.5 nátriumcsatorna szívizoformája. A közelmúltban számos mutációt írtak le ebben a fehérjében. Ezek megváltoztatják a Nav1.5 nátriumcsatorna biofizikai tulajdonságait in vitro, és az LQTS3-ban megfigyelthez hasonló fenotípust hoznak létre.28 Az esetek kevesebb mint 1%-át tulajdonítják ennek az oknak.
10-es típusú hosszú QT-szindróma (LQTS10)
A 10-es típust egy nagyon súlyos esetben írták le, QTc >600 ms, magzati bradycardia és 2:1 atrioventricularis (AV) blokk. A 11. kromoszómán (11q23) található SCN4B gén mutációi eredményezik, amely a nátriumcsatorna β4-alegységét kódolja. A β alegység négy különböző altípusát írták le, amelyek kölcsönhatásba lépnek és szabályozzák a különböző nátriumcsatorna-izoformákat; mindazonáltal eddig csak a 4-es altípust hozták összefüggésbe az aritmogenezissel.29 E gén mutációinak gyakoriságát nem vizsgálták, de becslések szerint
A Jervell-Lange-Nielsen-változat mutációi
Az LQTS e súlyos formáját az IKs áramot kódoló KCNQ1, és/vagy KCNE1 gének homozigóta30 vagy összetett heterozigóta mutációi okozzák; azaz az LQTS1 vagy LQTS5 formák nagyon súlyos változata. Ez az állapot jellemzően veleszületett süketséggel jár. A betegeknél általában QTc>500 ms és visszatérő syncope fordul elő, és nagy a hirtelen halál kockázata. Az ebben a változatban szenvedő betegek szülei általában heterozigóta, és kevésbé súlyos betegségben szenvednek, vagy nem mutatnak tüneteket.31
A HOSSZÚ QT-SYNDRÓMA DIAGNOSISA
Schwartz-pontszám
1985-ben Schwartz és munkatársai32 közzétették az LQTS diagnózisának kritériumait, amelyeket 1993-ban módosítottak, és fontos irányelveket tartalmaznak a lehetséges esetek kezdeti értékeléséhez. Ez a rendszer a családi anamnézis, valamint a klinikai és elektrokardiográfiás leletek alapján 1-től 9-ig terjedő pontszámot használ. A betegség valószínűsége ≥1 pontszám esetén alacsony, 2-3 pontszám esetén közepes, ≥4 pontszám esetén pedig magas (2. táblázat).

A hosszú QT-szindróma prenatális diagnózisa
A magzati bradycardia az LQTS egyik első klinikai manifesztációja lehet. Retrospektív sorozatok kimutatták, hogy a gyermekkorban LQTS-szel diagnosztizált betegek akár 70%-ánál előfordult bradycardia, általában magzati hydrops kíséretében.33 A magzati szív repolarizációjának értékelése a 14. és 39. hét között hasznos az LQTS korai diagnózisának felállításához.34
Az LQTS gonádális mozaikosságát a terhesség harmadik trimeszterében ismétlődő magzati veszteségekkel hozták összefüggésbe.35 A betegség erős gyanúja esetén a 16. terhességi hét után végzett amniocentézis hasznos lehet a diagnózis felállításában, amely könnyen elérhető, ha az egyik szülőről ismert, hogy egy specifikus mutáció hordozója.36
LONG QT-SYNDRÓMÁS BETEG Vizsgálata
Klinikai előzmények
Az LQTS diagnózisa és a kockázati rétegzés szempontjából egyaránt döntő jelentőségű a hirtelen halálozás családi és/vagy személyes előzményei. Ezenkívül a kiváltó tényezők és a syncope kontextusa jelezheti az LQTS altípusát. A gyanús eset kezdeti kiértékelése során ki kell zárni a QT-intervallumot meghosszabbító gyógyszerek használatát.
QT-intervallum:
A QT-intervallumot lehetőleg a II. vagy a V5-ös elvezetésekben kell mérni,37 ahol bizonyítottan nagyobb prediktív értékkel bír.38 Ez az intervallum a kamrai repolarizáció időtartamát jelzi, és a Q-hullám kezdetétől a T-hullám végéig mérik. Hagyományosan a Bazett39 által javasolt képletet alkalmazzák az intervallum időtartamának a pulzusszámnak megfelelő korrekciójára (QTc=QT/√RR, másodpercben kifejezve). Bár a QT-intervallum mérése egyszerűnek tűnik, a Viskin és munkatársai által végzett multicentrikus vizsgálatban40 a kardiológusokon kívüli orvosok kevesebb mint 40%-a, a kardiológusok kevesebb mint 50%-a és az aritmiával foglalkozó szakemberek több mint 80%-a tudta, hogyan kell helyesen mérni. Ajánlatos, hogy az orvosok manuális mérést végezzenek, és ne bízzanak az automatizált mérésekben, amelyek más intervallumok esetében hasznosak lehetnek, de a QT-intervallum kiszámításakor pontatlanok. A QT egy dinamikus intervallum, és a normális határértékek számos tényezőtől függenek. Bár férfiaknál az é440 ms, nőknél pedig az é460 ms QTc-intervallum kórosnak számít, ebben a tartományban találhatunk mutációk hordozóit és egészséges egyéneket is (3. ábra). Vincent és munkatársai41 LQTS1-ben szenvedő családokban kimutatták, hogy a pozitív genotípussal rendelkező esetek egyikében sem volt QTc470 ms. Monnig és munkatársai38 nemrégiben kimutatták, hogy a QTc>440 ms elegendő az LQTS-hez társuló mutációkkal rendelkező betegek kimutatására, a QTc>470 ms hasznos a tünetek kialakulásának kockázatának kitett betegek azonosítására, a QTc>500 ms pedig a kezelés alatt álló tünetes betegeknél található.
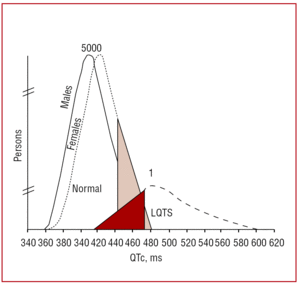
3. ábra. A szívfrekvenciával korrigált QT-intervallum (QTc) eloszlását bemutató modell a KVLQT1, HERG vagy SCN5A mutációval rendelkező betegek és nem érintett családtagjaik esetében. A bal oldali görbe az érintetlen, a jobb oldali görbe pedig az érintett családtagok eloszlását írja le.
A hosszú QT-szindrómához társuló egyéb elektrokardiográfiás elváltozások
Az LQTS-ben szenvedő betegek többféle T-hullámváltozást is mutathatnak: többek között polaritásváltozást, amplitúdóváltozást, bevágást és kétfázisú megjelenést.42 A T-hullám alternans (4A. ábra) a szinuszritmusú T-hullám amplitúdójának, morfológiájának és polaritásának ütemenkénti változása, a QRS-komplexus változása nélkül. Ez az elektromos instabilitás indikátora,43 a kamrai repolarizáció regionális szóródását tükrözi, és esetenként megelőzi a kamrafibrillációt.44
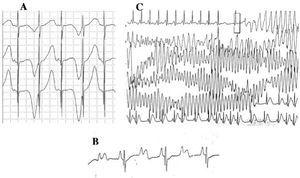
4. ábra. Elektrokardiográfiás elváltozások hosszú QT-szindrómában. A: T-hullám elektromos alternancia. B: 2:1 arányú atrioventrikuláris blokk. C: önkorlátozó torsade de pointes.
Az LQTS-ben szenvedő betegeknél a szinuszcsomó diszfunkciójának jelei, bradycardia és/vagy szünetek jelentkezhetnek.45 Az LQTS1 és LQTS3 altípusok, különösen az utóbbi, gyakran szinuszbradycardiát mutatnak,46 míg az LQTS4-hez szinuszcsomó-diszfunkciót társítottak.18
Az 1970-1980-as évtized óta megfigyelték az AV-vezetési hibák és az LQTS47 együttes előfordulását (4B ábra). A kettő-egy AV-blokk egy ritkán előforduló, rossz prognózissal járó megnyilvánulás, amely már a magzati stádium óta jelen lehet perzisztáló bradycardia formájában. Ennek a rendellenességnek az előfordulási gyakorisága 4-5%48 , és a béta-blokkolókkal és/vagy pacemakerrel történő kezelés ellenére magas mortalitással jár.49,50 Ez a jelenség az akciós potenciál hosszú időtartamával magyarázható. Ha a kamrai refrakter periódus meghosszabbodik, a szinuszaktivitás következő impulzusa blokkolódik, mert akkor éri el a kamrákat, amikor azok még a refrakter periódusban vannak. Úgy tűnik, hogy ez a változás kizárólag LQTS-ben fordul elő, mivel a kamrai refrakter periódus nagyobb, mint az AV-vezetésé.51 A QRS-komplex lejtése általában meredek, és a blokkot az infraHis területen lokalizálták,46,51,52 de a blokk helye függhet a genotípustól. Eddig 4 gént hoztak összefüggésbe az LQTS 2:1 blokkjával: HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 és SCN4B (LQTS10).55
Az LQTS jellegzetes kamrai aritmiája az úgynevezett torsade de pointes (4C ábra). Ez akkor jelentkezik, ha a QT-intervallum meghosszabbodik, függetlenül az etiológiától. Ez egy reentry okozta polimorf kamrai tachycardia, amelyet elektrokardiográfiailag a QRS-tengely folyamatos csavarodása jellemez egy képzeletbeli vonal körül. Általában szünet előzi meg, amelyet extrasystolé követ (rövid-hosszú-rövid RR-intervallum), ahogy az ábrán látható.56-58 Kamrafibrillációban és hirtelen halálban csúcsosodhat ki. Ha ez nem következik be, a beteg csak szinkópát tapasztalhat, és ha az epizód rövid, észrevétlen maradhat.
Holter
A Holter-vizsgálat a QT-intervallum teljes körű, dinamikus értékelését teszi lehetővé. Esetenként rögzítik a tünetmentes kamrai ritmuszavar spontán epizódjait, valamint a sinuscsomó diszfunkció vagy AV-blokk epizódjait.
Terheléses terheléses vizsgálat
Az LQTS-ben szenvedő betegek nem tudják elérni az életkoruknak megfelelően számított maximális várható pulzusszámot. Emellett terhelés alatt a QT-intervallum paradox viselkedést mutathat, ahelyett, hogy csökkenne, inkább növekszik.59,60 A terheléses terheléses vizsgálat során az elektrokardiográfiás mintázat az LQTS típusától függően eltérő lesz. Az LQTS1-es betegek amellett, hogy nem érik el az életkoruknak megfelelő maximális számított pulzusszámot, gyakran megnövekedett QT-intervallumot mutatnak, míg az LQTS2-es betegek elérhetik az elvárt pulzusszámot, és csak enyhe QT-intervallum-növekedést vagy egyáltalán nem mutatnak.61,62 Általában az LQTS3-as betegeknél a terhelésre adott fiziológiás válasz, azaz a QT-intervallum normális rövidülése figyelhető meg.63 A terheléses vizsgálat hasznos lehet a kezelésre adott válasz értékelésére és a kockázat rétegzésére is tünetmentes esetekben, vagy ha kétségek merülnek fel az aritmiához vezető eseményekkel kapcsolatban.
Genetikai szűrés
Az utóbbi években az LQTS genetikai vizsgálatai a kutatólaboratóriumokra korlátozódtak. Mindazonáltal az ezekből az erőfeszítésekből származó információk rendkívül hasznosak voltak a betegek, különösen a nagy kockázatú esetek kezelésében. A szűrés talán fő alkalmazási területe a genetikai tanácsadás, de fontos következményei vannak a kezelésben is, amelyet az érintett csatornának megfelelően lehet orientálni. Az adott mutáció pontos elhelyezkedése további információkkal szolgálhat a kockázat alakulását illetően. A KCNQ1 transzmembrán régiójában (IKS) mutációval rendelkező betegeknél nagyobb valószínűséggel fordulnak elő aritmiás események, mint a C-terminális régióban64; ugyanez igaz a KCNH2 vagy a HERG pórus régiójában65 mutációval rendelkező betegeknél az N- vagy C-terminális régióban mutációval rendelkező betegekhez képest.66
A kezdeti szűrés talán a KCNQ1, HERG és SCN5A génekre korlátozódhat, amelyek az esetek 65%-ában lehetőséget adnak a mutációkkal való találkozásra. Ha a kapott eredmények negatívak, a szűrés kiterjeszthető a KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 és SCN4B génekre, ami 5-10%-kal növeli a pozitív eredmények lehetőségét.
Postmortem genetikai szűrés
Érdekes, hogy LQTS-hez vezető génmutációkat találtak olyan gyermekeknél, akiknél hirtelen halál következett be, valamint fiatal felnőtteknél hirtelen halál megmagyarázhatatlan eseteiben.
Hirtelen halálesetet szenvedett és negatív boncolású betegek postmortem genetikai vizsgálatai különböző százalékban mutattak ki LQTS-hez vezető mutációkat67-69: gyermekeknél közel 10%, fiatal felnőtteknél 35%.70-72 Ezen eredmények alapján javasolták a rutinszerű EKG-vizsgálatot minden újszülöttnél.73,74
A postmortem genetikai vizsgálat, amelyet az irodalomban “molekuláris autopsziának” is neveznek, a jogi következményeken túlmenően fontos következményekkel jár a családokban, akik anélkül lehetnek érintettek, hogy tudnának róla.
Szabályozó polimorfizmusok
Az LQTS populációban számos gyakran előforduló polimorfizmust írtak le, amelyek szinte valamennyi, az állapothoz kapcsolódó génben eloszlanak. Bár ezek a változások nyilvánvalóan nem patogén jellegűek, némelyiküknek a következő hatásai lehetnek75-78:
1. Egyéni fogékonyságot generálnak az aritmia kialakulására.
2. Kedvezik egy másik nem szinonim változás patogén hatását.
3. Csökkentik egy másik nem szinonim változás patogén hatását.
Ilyen a KCNH2 (HERG) K897T polimorfizmusa, amely a populáció akár 15%-ánál is előfordul, és nemcsak bizonyos gyógyszerekre való fogékonysággal van összefüggésben,79 hanem az ugyanabban a génben lévő mutációk patogén hatását is kedvezően befolyásolja.78 Egy másik példa az SCN5A gén S1103Y polimorfizmusa, amely főként feketéknél fordul elő, előfordulása közel 13%-os, és a gyermekkori hirtelen halál fokozott kockázatával jár együtt80.
Érdekes módon az SCN5A gén termékében (amely az emberben a Nav1.5 nátriumcsatorna izoformáját kódolja) két alternatív feldolgozási helyet írtak le, amelyek kétféle nátriumcsatornát generálnak: az egyik 2016 aminosavat tartalmaz glutamint az 1077-es pozícióban (Q1077), a másik 2015 aminosavat, amelyből hiányzik a glutamin (Q1077del). Ezen alternatív feldolgozások transzkriptumai 2:1 arányban vannak jelen ugyanabban az emberi szívben, és számos gyakori polimorfizmus különböző hatással lesz a csatorna működésére, attól függően, hogy a kontextus Q1077 vagy Q1077del. Ezt először az SCN5A H558R polimorfizmusával mutatták ki, amely a populáció akár 30%-ában is jelen van. Amikor a H558R-t Q1077 kontextusban fejezték ki, az ionáram mélyreható csökkenését figyelték meg.81 Hasonló hatást dokumentáltak az S524Y82 polimorfizmus esetében is. Ezek az eredmények magyarázó tényezőkkel szolgáltak a betegség eltérő súlyosságára, valamint az egyes családokban megfigyelt eltérő fenotípusokra ugyanazon mutáció esetén.77
Farmakológiai vizsgálat adrenalinnal
A kis dózisú adrenalinnal végzett farmakológiai vizsgálat biztonságos és hasznos lehetőség az LQTS feltételezett, határes QTc-jű eseteinek leleplezésére. Különösen hatékony az LQTS1 tünetmentes formáinak kimutatására, 92,5%-os érzékenységgel, 86%-os specificitással, 76%-os pozitív prediktív értékkel és 96%-os negatív prediktív értékkel. Hasznos lehet az LQTS2 diagnózisában is, alacsonyabb érzékenységgel és specificitással. Nem hasznos az LQTS3 vagy az LQTS egyéb formái esetében. Normális körülmények között a szimpatikus ingerlés az IKs káliumcsatorna foszforilációját indukálja, optimalizálva annak működését és az akciós potenciál rövidülését eredményezve. Az LQTS-ben, különösen az 1-es típusban szenvedő betegeknél paradox reakció figyelhető meg kis dózisú adrenalin (0,025-0,2 µg/kg/min) adására, amely a QT-intervallumot több mint 30 ms83-86-ra meghosszabbítja.
A QT-intervallum meghosszabbodása és a gyógyszerek által kiváltott TORSADE DE POINTES
A különböző orvosi szakterületeken alkalmazott gyógyszerek nagy száma okozhatja a QT-intervallum iatrogén megnövekedését. Egyes gyógyszereket e nemkívánatos hatás miatt kivontak a forgalomból (pl. többek között az asztemizol és a ciszaprid; további információért látogasson el a www.qtdrugs.org oldalra).87,88
A nem antiaritmiás gyógyszerek által okozott másodlagos kamrai aritmia 10 000-100 000 exponált személy közül kevesebb mint egynél fordul elő. Tekintettel arra, hogy a klinikai vizsgálatok 2000-3000 alanyra terjednek ki, ez a nemkívánatos és halálos kimenetelű mellékhatás a gyógyszerfejlesztés klinikai szakaszában könnyen elkerülné a felderítést.89 Ez a pont óriási érdeklődést váltott ki az új gyógyszerek tanulmányozása és fejlesztése során a biztonságosságra vonatkozó szempontok iránt.
Az egyéni fogékonysággal kapcsolatos tényezők közé tartozik a női nem, a hipokalcémia, a hipomagnezémia, a bradikardia, a szívelégtelenség, a posztkardioverzió, a pitvarfibrilláció, a bal kamrai hipertrófia, a fel nem fedezett LQTS, a hajlamosító polimorfizmusok és a hajlamosító gyógyszerek magas szérumkoncentrációja.90
A gyógyszerekkel jellemzően kölcsönhatásba lépő csatorna molekuláris szerkezete miatt a KCNH2(HERG) gén által kódolt IKr. Más káliumcsatornák 2 prolin-maradványa a csatorna pórusa felé van szögelve, csökkentve annak lumenét. Ezzel szemben az IKr-ből hiányoznak ezek a maradékok, nagyobb pórus előtere keletkezik, és megkönnyíti a nagy molekulák expozícióját. Ezenkívül 2 aromás maradékkal rendelkezik (tirozin és fenilalanin), amelyek kedveznek a csatornát blokkolni képes számos gyógyszerben jelen lévő aromás molekulákhoz való kötődésnek.91
Mint már említettük, az LQTS penetrációja nem teljes, és a mutációk egyes tünetmentes hordozói rosszindulatú aritmiát manifesztálhatnak, ha valamelyik ilyen gyógyszert kapják. Ezenkívül a populációban gyakorinak tartott polimorfizmusok egyes gyógyszerek alkalmazása esetén egyéni fogékonyságot kölcsönöznek a torsade de pointes kialakulására. Ez a helyzet az R1047L polimorfizmus esetében, amely a második leggyakoribb a KCNH2 génben, és amelyet a dofetilid nevű gyógyszer alkalmazásakor torsade de pointes kialakulásával hoztak összefüggésbe.92 Legalább 20 KCNH2 génpolimorfizmust írtak le egészséges személyeknél, és még meg kell határozni a gyógyszerekkel kapcsolatos aritmiára való egyéni fogékonyságra gyakorolt hatásukat.93 A Na1.5 nátriumcsatornában is dokumentáltak a kamrai aritmia kialakulására való fogékonyságot adó polimorfizmusokat. Ilyen a H558R polimorfizmus, amely a populáció akár 30%-ánál is előfordul, vagy az S1103Y, amely a feketéknél gyakori80,81,90,94,95; a gyógyszerek okozta fogékonyságban betöltött szerepüket nem vizsgálták.
LONG QT SYNDROMA ÉS TERHESSÉG
A genetikai tanácsadás fontos az LQTS esetében, de általánosságban elmondható, hogy a terhesség nem ellenjavallt a hordozó nők esetében, bár minden eset más és más, és egyénileg kell értékelni a megfelelő összefüggésben.
Megállapították, hogy a rosszindulatú kamrai aritmia kialakulásának kockázata csökken a terhességgel. Ezzel szemben a szülés utáni első 9 hónapban nagyobb veszélyeztetettségről számoltak be a malignus aritmia megjelenése szempontjából, különösen az LQTS2-ben szenvedő betegek esetében. Ez a kockázat béta-blokkoló kezelés hatására jelentősen csökken.96
RIZIKÓ STRATIFIKÁCIÓ
Az LQTS alakulása változó, és befolyásolja a QTc-intervallum időtartama, a környezeti tényezők, az életkor, a genotípus és a kezelésre adott válasz.97,98 A kamrai ritmuszavar gyakrabban fordul elő LQTS1 és LQTS2-ben, de súlyosabb az LQTS3-ban.99 Mint már említettük, a nők különösen fogékonyak a rosszindulatú ritmuszavarra a szülés utáni időszakban.14
A hosszú QT-szindrómát akkor kell magas kockázatúnak tekinteni, ha az alábbiakkal társul:
1. A hosszú QT-szindróma a következő tünetekkel jár:
1. A szülés utáni időszakban a nők a szülés utáni időszakban különösen fogékonyak a rosszindulatú ritmuszavarra. Veleszületett siketség (Jervell-Lange-Nielsen-szindróma).
2. Rosszindulatú kamrai tachyarrhythmia miatt visszatérő syncope.
3. Hirtelen halálozás a családban.
4. Hirtelen halál a családban. QTc>500 ms.
5. 2:1 arányú atrioventrikuláris blokk.
6. Szívritmuszavar. T-hullám elektromos alternansz.
7. LQTS3 genotípus.
A Priori és munkatársai97 647 betegen végzett vizsgálata kimutatta, hogy a 40 éves kor előtti súlyos esemény (syncope, szívmegállás, hirtelen halál) előfordulásának valószínűsége magas (>50%), ha a QTc >500 ms LQTS1, LQTS2 és LQTS3-ban szenvedő férfiaknál. Nemrégiben a nemzetközi LQTS-regiszter elemzéséről számoltak be. A hirtelen halálozás kockázatát 2772, a betegségben szenvedő serdülőnél elemezték, és 3 olyan tényezőt azonosítottak, amely ebben a populációban magasabb kockázattal jár: A 10-12 éves fiúknál magasabb volt a kockázat, mint a lányoknál, de a 13-20 éves korosztályban a kockázat hasonló volt.100
KEZELÉS
A kezelésben nem részesülő tünetes betegeknél az első kamrai aritmiás eseményt követően az éves halálozási arány 20%, a 10 éves halálozás pedig 50%. Bár egyértelmű, hogy a kezelést a tünetek jelentkezésekor kell megkezdeni, a tünetmentes betegeknél alkalmazandó megközelítés még mindig vita tárgyát képezi. Dokumentálták, hogy a betegek 9%-ánál a szívmegállás lehet a betegség első megnyilvánulása,48 és hogy a tünetmentes betegek 12%-ánál jelentkeznek tünetek, és előfordulhat hirtelen halál. A béta-blokkolókkal való kezdeti kezelést minden LQTS-es betegnél meg kell kezdeni. A testmozgás korlátozása ajánlott, de a klinikai és elektrokardiográfiás kockázati markerek hasznos alapot jelentenek a döntéshozatalhoz. Fontos, hogy a betegeket tájékoztassuk több olyan gyógyszer alkalmazásának kockázatáról, amelyek meghosszabbíthatják a QT-intervallumot és kedvezhetnek a kamrai aritmia kialakulásának, ahogyan azt fentebb említettük. A genetikai diagnózis, amellett, hogy lehetővé teszi a betegséggel kapcsolatos megfelelő családi tanácsadást, segítséget nyújt a prognózis megítélésében és a specifikus kezelés orientálásában.
Béta-blokkolók
A béta-blokkolók jelentik az LQTS első vonalbeli kezelését, és minden betegnek ezeket kell kapnia kezdeti terápiaként.101 Akár 64%-kal100 is csökkentik a kardiovaszkuláris események kockázatát, és különösen hatékonyak az IKs csatorna mutációban (LQTS1) szenvedő betegeknél,102 amelyeket nagymértékben a szimpatikus rendszer szabályoz. A béta-blokkolók nem a QT-intervallumot, hanem annak szóródását módosítják.103 Bár ezek a gyógyszerek csökkentik az események előfordulását,104,105 kimutatták, hogy az LQTS1-es betegek 10%-ánál, az LQTS2-es betegek 23%-ánál és az LQTS3-as betegek 32%-ánál a kezelés ellenére is jelentkeznek kardiovaszkuláris tünetek.106 Úgy tűnik, hogy különösen az LQTS3-ban szenvedő betegek nem érnek el jelentős előnyöket; valójában ezt a gyógyszercsoportot óvatosan kell alkalmazni ezeknél a betegeknél, mivel az LQTS3-ban a kamrai aritmia epizódjai gyakrabban fordulnak elő, amikor a szívfrekvencia alacsony. Általánosságban elmondható, hogy a tüneteket mutató betegek 32%-ánál a béta-blokkoló kezelés megkezdése előtti első 5 évben ismétlődő tünetek jelentkeznek, és a hirtelen halálos epizódból megmenekült betegek 14%-ánál 5 éven belül újabb hasonló esemény jelentkezik, ha csak ezt a terápiát kapják.107 Az LQTS kezelésében számos béta-blokkolót alkalmaznak, elsősorban nadololt (0,5-1 mg/kg/nap), propranololt (2-4 mg/kg/nap), metoprololt (0,5-1 mg/kg/nap) és atenololt (0,5-1 mg/kg/nap). Az atenolol azonban nem feltétlenül előnyös LQTS-ben; közölték, hogy a béta-blokkoló kezelésre nem reagáló betegek legalább 75%-a atenololt kapott, bár ez a megállapítás összefügghet a szuboptimális dózisok alkalmazásával.104 A megfelelő dózis megállapításához hasznos a terheléses vizsgálat. A maximális pulzusszám nem haladhatja meg a 130 ütés/perc értéket a kezelés alatt.
Nátriumcsatorna-blokkolók
Az LQTS3-t okozó nátriumcsatorna-mutációk a csatorna hibás inaktivációját eredményezik; a nátriumcsatorna-blokkolás hasznosnak bizonyult ezeknél a betegeknél. A flekainiddal végzett vizsgálatok a szívfrekvencia, a T-hullám-változások és a QT-intervallum javulását dokumentálták.108 A mexiletin szintén javítja az elektrokardiográfiás kockázati markereket.63,109,110 A ranolazinnal végzett in vitro vizsgálatok az emberekben jelentett mutációk káros hatásainak csökkenését mutatták.111 Bár az eredmények biztatóak, szem előtt kell tartani, hogy nincsenek hosszú távú, ezt a terápiát értékelő vizsgálatok, és nagy sorozatból származó eredményekről sem számoltak be. Nátriumcsatorna-blokkolók nem adhatók, ha nincs megerősített genetikai diagnózis.
Káliumpótlás és a rendelkezésre állását növelő gyógyszerek
A káliumpótlás és/vagy a káliumkímélő gyógyszerek, mint például a spironolakton, az esetek 24%-ában rövidítik a QTc-intervallumot.112,113 A káliumcsatornák megnyitását elősegítő gyógyszerek, mint az aprikalim, levcromakalim, nikorandil és pinacidil, hasznosnak bizonyultak az LQTS kezelésében. Az altípusok, amelyekben különösen előnyösek, az LQTS1 és az LQTS2.114
Pacemakerek és defibrillátorok
A pacemaker-stimulációt pauzafüggő aritmiában szenvedő betegeknél alkalmazták.115,116 Az LQTS3-ban szenvedő betegek általában jobban profitálnak ebből a kezelésből, mivel a bradycardia gyakorisága nagyobb ebben a csoportban. A DDD pacemelés szünetfüggő ritmuszavarban vagy magas fokú 2:1 AV-blokkban szenvedő betegeknél javallott. A 70 ütem/perc117 alá programozott frekvenciák nem hasznosak a kamrai aritmia megelőzésére. Ajánlott az érzékelőt gyors válaszra programozni, mivel ezeknél a betegeknél általában nem megfelelő szívfrekvencia-gyorsulás jelentkezik terhelés hatására. Minden olyan funkciót, amely szünetek jelenlétét feltételezi, ki kell kapcsolni, mint például a hiszterézis és az éjszakai funkció. A PARP-nek (posztventrikuláris pitvari refrakter periódus) a lehető legrövidebbnek kell lennie. A frekvenciaszabályozó funkciót be kell kapcsolni a posztextraszisztolés szünet megelőzése érdekében. Nem szabad elfelejteni, hogy a T-hullám túlérzékelése és a befogási hibák is okozhatnak szüneteket. A beültethető kardioverter defibrillátor (ICD) és béta-blokkolók kombinált alkalmazása jelentősen csökkenti a hirtelen halál előfordulását.118-120 Ezen intézkedések indikációja egyértelmű a magas kockázatú esetekben.121 Az eszköz programozása az egyes betegek igényei szerint változik, de általában kerülni kell a kezelés beadását tünetmentes, önkorlátozó eseményeknél; ennek érdekében 15 s-os detektálási idő indikált. Az aritmiás vihar az AID-terápia szövődménye. A betegek közel 15%-ánál fordulhat elő ez a szövődmény, amely jórészt az ICD-sokkot követő fokozott szimpatikus tónusnak köszönhető.118 Ez a probléma a béta-blokkoló adagjának növelésével kezelhető. Ha ez az intézkedés nem célravezető, a szimpatikus lánc ganglionjainak reszekcióját kell megfontolni.
Bal szimpatektómia
1971-ben a szimpatikus gangliektómiát mint hasznos terápiás lehetőséget mutatták be ezekben a betegekben.122 1991-ben Schwartz és munkatársai123 publikálták az első sorozatot 85, a béta-blokkoló kezelésre rosszul reagáló betegről, akiknél bal szimpatikus gangliektómiát végeztek, biztató eredményekkel: az 5 éves túlélés aránya 94% volt. Jelenleg ezt a terápiás lehetőséget azoknak a nagy kockázatú betegeknek ajánlják, akiknél a béta-blokkoló kezelés és/vagy a pacemaker beültetés ellenére is fennáll a syncope, valamint azoknak, akiknél a beültetett defibrillátor gyakori sokkot okoz. Az eljárás a stellatus ganglion inferior részének és a szimpatikus lánc bal oldali T2-T4-es mellkasi ganglionjainak reszekciójából áll, mivel az egyszerű bal oldali stellektómia nem bizonyult kellően hatékonynak. A mikroinvazív thorakoszkópiát124,125 jó eredménnyel alkalmazták. Az ezzel a módszerrel kezelt betegek legnagyobb sorozatáról nemrégiben számoltak be, amely a syncope-epizódok vagy a hirtelen halálesetek számának jelentős csökkenését, valamint 95%-os 5 éves túlélési arányt mutatott. A korábbi syncopéval rendelkező betegek esetében az 5 éves túlélés 97% volt, a kiújulás lehetősége 11% volt, amely a legtöbb esetben egyetlen syncopéból állt. A bal oldali szimpatektómiát követően a QT-szakasz jelentős csökkenése is megfigyelhető volt. E kedvező eredmények ellenére a hirtelen halál megelőzése nem teljes, de 3%-ra csökkent. Az ICD-vel rendelkező betegeknél, akiket többszörös defibrillátor-sokkok miatt műtöttek meg, az események átlagos száma 25-ről 0-ra csökkent, ami 95%-os csökkenést jelent. A kedvező hatást megerősítették az LQTS1 esetében. Az LQTS2-ben szenvedő betegeknél az előnyök valószínűleg kisebbek, LQTS3-ban pedig nem bizonyított a hatékonysága.126
Abláció
Beszámoltak arról, hogy a kamrai aritmiát egyes esetekben kiváltó extrasystolé ablációja az epizódok előfordulásának csökkenésével végezhető.127 Nincsenek azonban megfelelő számú betegen végzett hosszú távú vizsgálatok, amelyek indokolnák e technika rutinszerű alkalmazását.
Lásd a 675-82. oldalon található vezércikket
LEÍRÁSOK
AV: atrioventricularis
AID: automatikus beültethető defibrillátor
ECG: elektrokardiogram
QTc: szívfrekvenciával korrigált QT
ATS: Andersen-Tawil szindróma
LQTS: hosszú QT szindróma
Dr. Medeiros gazdasági támogatást kap a CONACyT-től és a FUNSALUD-tól.
.