INTRODUCTION
QT延長症候群(LQTS)は、心室再分極が大きく変化し、心電図上のQT間隔が延長することが特徴である。 この疾患は,悪性心室性不整脈(torsade de pointes)や突然死の素因となる。 QT 延長症候群の臨床的および心電図的記述は、1957 年に Anton Jervell と Fred Lange Nielsen1 によって報告され、非血縁の両親と 6 人の子供の家族に関する研究が発表された。 このうち4人の子供には先天性難聴と失神エピソードがあり、3人は突然死を呈していた。 これらの患者の心電図検査では、異常に長いQT間隔が認められた。 両親とも無症状で、心電図も正常であり、聴覚障害もなかった。 1964年、RomanoとWardは、神経性難聴を伴わない再発性失神、突然死の家族歴、QT間隔の延長を特徴とする心臓症候群を独立して報告した2。その後の遺伝子研究により、JervellとLange Nielsenが述べた先天性神経難聴を伴う症候群は、ホモ接合体変異に対応し、表現型は重篤で突然死の危険性も高いことが明らかにされた。 ロマノ・ワード症候群として知られる疾患は、一般にヘテロ接合体の変異に対応し、患者は聴覚の変化を示さず、その重症度はかなり異なる。 それから約半世紀後の1995年、LQTSの主要な関連遺伝子が報告され3,4、この疾患は心臓のイオンチャンネル異常であると認識されるようになった。 LQTSは、初めて報告された心臓のチャネル異常症であり、おそらく今日までで最も広く研究されている不整脈誘発性イオンチャネル疾患である。 臨床像は実にさまざまで、無症状の場合もあれば、失神、発作、突然死などを繰り返しながら進行する場合もある。 当初、LQTSは稀な症候群と考えられており、事実上、重篤な症状の発現は散発的である。 しかし、関連する変異の発生率は1/3000-5000と推定されており5、無症候性保因者の32%は心拍補正したQT間隔(QTc)が正常範囲内にあることがあり、その子孫の50%がこの疾患を発症し、一般集団と比較して不整脈を起こしやすく、最大20%が症状を呈することがある6
QT延長症候群には大きな遺伝的異質性が認められる。 この疾患では、10の遺伝子に分布する500以上の変異が報告されている。 KCNQ1、HERG、SCN5A、KCNE1、KCNE2、ANKB、KCNJ2、CACNA1、CAV3、SCN4Bの10遺伝子に分布する500以上の変異が報告されている。 この分野の進歩にもかかわらず、25%~30%の患者では遺伝子診断を確立することができません。7,8 この疾患の症状は主に単発性で6、多発性または複合型は通常より重篤な表現型となります。 ペネトランス(変異を有し、その表現型を示す患者)は25%から90%である9。あまり多くはないが、疾患の発現率に変動があり、同じ変異から複数の表現型が生じることがある。 過去11年間の分子遺伝学的研究により、遺伝子型と表現型の重要な相関が明らかになり、治療法の指針となっています。 さらに、この集団で頻繁に見られる非同義多型を調査した結果、不整脈の発症に対する個人の感受性について興味深い観察がなされており、特にファーマコゲノミクスの分野で大きな関心を呼んでいる。
Long QT症候群の分類
General Concepts
過去に用いられたLQTSの分類は、ホモ接合体かヘテロ接合体かで、それぞれJervell-Lange-Nielsen症候群(難聴あり)、Romano-Ward症候群(難聴なし)という病態を生じている。 本分類では、表1に示すように、遺伝的所見を重視した分類を行っている。 本疾患に関連する3つの主要な遺伝子は、1995年から1996年にかけて記述された。 これらの遺伝子は、カリウムチャネルIKsとIKrの孔形成ユニット、およびナトリウムチャネルNav1.5をコードしており、症例の65%近くを占めている。
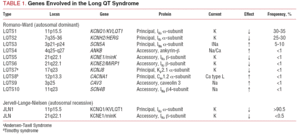
イオンチャネルは、細胞膜を通してイオンを輸送する膜貫通型タンパク質であり、その役割は、細胞膜を貫通してイオンを輸送することである。 LQTSに関与するチャネルは、単一のイオンを輸送することに選択的または特化しており、電圧依存性である、すなわち、チャネルのサブタイプによって異なる特定の細胞内電圧で活性化される。 心筋細胞で起こる電気的現象や収縮現象は、これらの構造によって制御されている。 イオンチャネルは、チャネル孔を形成するメインユニットとそれを制御する補助的なタンパク質からなる高分子複合体を形成している(図1)。 LQTSで見られるチャネルの機能障害は、この主タンパク質と制御タンパク質の2つの部位で発生する可能性がある(表1)。 αとして知られる孔形成ユニットの関与は、LQTSの最も一般的な3つの亜型を生み出す。 LQTS1(IKsカリウムチャネルに影響)、LQTS2(IKrカリウムチャネルに影響)、LQTS3(ナトリウムチャネルに影響)である。 これらは最も頻度の高い亜型であるため、臨床的にも遺伝学的にも最もよく特徴づけられている。 これらの3つの主な型における表現型と遺伝子型の相関を図2に示す。 現在、Jervell-Lange-Nielsen症候群はLQTS 1型と5型に相当する。 これらの患者は先天性難聴とIKs電流に影響を与える複合ホモ接合体またはヘテロ接合体変異を持つことが特徴である。 Romano-Ward症候群はLQTS 1から10までを含み、難聴を伴わない。
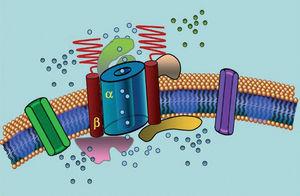
図1. 高分子複合体の模式図。 イオンチャネルは様々なタンパク質によって制御される膜貫通タンパク質(α)であり、その1つがいわゆるβサブユニットである。
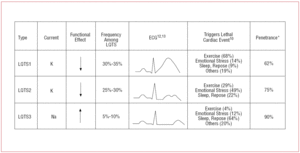
図2.活動電位が延長されるメカニズム 最も頻度の高いQT延長症候群における遺伝子型-表現型の相関。 *LQTS2 の患者は、感情的ストレス (49%) や突然の聴覚刺激 (例: 目覚まし時計) に反応して心室性不整脈を示す傾向があり、睡眠中 (22%) や運動中 (29%) は少ない。10 産後の女性は特に影響を受けやすい14。 推定浸透率は79%である。したがって、最大で20%の症例が診断不能な心電図を示す可能性がある。 LQTS2のT波は通常低振幅で2分し、ノッチングを伴う12,13 (図2)。 患部遺伝子はKCNH2またはHERGで、第7染色体(7q35-36)上にあり、IKrカリウムチャネルα-サブユニットをコードし、症例の25%〜30%を占める。 このチャネルの機能不全は、活動電位の第3相の間に出て行くK+電流を減少させ、その持続時間を延長させる。
QT延長症候群3型(LQTS3)
LQTS3型の患者は、安静時(睡眠時)や徐脈時に悪性不整脈を呈するリスクが高い15。SCN5A遺伝子変異の浸透度はほぼ90%である。 LQTS3の心電図は通常、遅延した尖ったT波を示し、STセグメント延長を明確に観察することができる12,13(図2)。 LQTS3の原因遺伝子は、第3染色体(3p21-24)上のNav1.5ナトリウムチャネルαサブユニット(図1)をコードするSCN5Aで、5~10%の症例で発症する。 このチャネルの不活性化不全により、活動電位の第2相にNa+が持続的に入力され、その持続時間が延長する。
QT延長症候群4型(LQTS4)
4型はLQTSのまれな型で、1%近くを占める。 カテコールアミン作動性多形性心室頻拍、心房細動、心室内伝導障害、洞房結節機能障害、徐脈など幅広い不整脈を生じる非定型型であり、また、多くの患者でQTcは正常範囲内であることがある。 この遺伝子は、心筋細胞の膜タンパク質と細胞骨格タンパク質をつなぐ構造タンパク質であるアンキリン-βの合成をコードしています。 これらのタンパク質は、Na/K ATPaseポンプ、Na/Ca交換体、イノシトール三リン酸受容体(InsP3R)である。 アンキリンβの機能が失われた変異体では、細胞内カルシウム濃度が上昇し、N/K ATPaseやNa/Ca交換体の発現に変化が生じる。 カルシウム濃度の上昇は、早期および遅発性後脱分極を引き起こす。 したがって、アンキリンβ遺伝子変異で観察される心室性不整脈は、通常カテコールアミン作動性刺激に応答した自発的脱分極に起因している。
5型QT症候群(LQTS5)
5型は、21番染色体(21q22.1p22)にあるKCNE1遺伝子の配列の変化に由来し、KCNE1はIKsチャネルβ-サブユニット(minKサブユニットとしても知られ)を制御する合成をコードしている19。 この型は症例の1%未満である。
6型QT症候群(LQTS6)
6型の影響遺伝子は、21番染色体(21q22.1)にあるKCNE2であり、20 この遺伝子は、カリウムチャネルβ-サブユニット、またMiRP1サブユニットとして知られ、IKrチャネルを調節することをコード化する。 QT 延長症候群 7 型または Andersen-Tawil 症候群 (LQTS7)
この症候群で見られる異形所見と心電図変化は、1971 年に Andersen 博士21 によって初めて報告され、1994 年に Tawil 博士22 によって再検討されたが、遺伝学/分子学的な説明は 2001 年に報告されるまでなされなかった。23 現在、Andersen-Tawil症候群(ATS)として知られているこの疾患は、周期性麻痺、骨格形成異常、心室性期外収縮を頻繁に伴うタイプの心室性不整脈、および特に女性における心室細動の発症しやすさを特徴とする常染色体優性遺伝の変異体である。 ATSで報告されている変化は、心室性期外収縮(41%)、非持続性多形心室頻拍(23%)、双方向性心室頻拍(68%)、Torsade de Pointes(3%)です。24 観察される異形の特徴には、低身長、側湾、臨床指腸、多指腸、低埋没耳、小顎、広い額などが含まれます。 23,25 17番染色体(17q23)に位置するKCNJ2遺伝子の変異は、整流カリウムチャネルKir 2.1の合成をコードし、70%の症例で見られる。 このチャネルは活動電位の第4相に関与している。 この症候群ではQTc間隔がわずかに延長するだけか、あるいは正常でもU波が目立つため、QT間隔が過大評価されていることから、この遺伝子をLQTSの原因遺伝子に含めることに疑問を呈する著者もいる。 24
8型QT延長症候群(LQTS8)
8型は、L型カルシウムチャネルCav1をコードする12番染色体(12p13.3)上のCACNA1遺伝子に変異が生じることにより発症する。心奇形、間欠性免疫不全、低血糖、自閉症などの認知障害、指間融合、不整脈や突然死を引き起こすQT延長などを特徴とするTimothy症候群26の原因となっている27。 8型は0.5%未満である。
9型QT症候群(LQTS9)
この種のLQTSは、第3染色体(3p25)にあるカベオリン3合成をコードするCAV3遺伝子に変異が生じることで発症する。 カベオラはエンドサイトーシス、脂質の恒常性、シグナル伝達に関与する細胞膜の侵襲である。 この構造の重要な構成要素はカベオリンであり、3つのサブタイプが知られており、サブタイプ3は骨格筋と心筋に特異的である。 カベオラには、ナトリウムチャネルNav1.5の心筋アイソフォームを含むいくつかのイオンチャネルが併存している。 このタンパク質のいくつかの変異が最近報告されている。 LQTS10型は、QTc >600ms、胎児性徐脈、2:1房室ブロックを伴う非常に重症の症例で報告されている。 11番染色体(11q23)上に位置し,ナトリウムチャネルβ4-サブユニットをコードするSCN4B遺伝子の変異に起因する。 4種類のβ4サブユニットが報告されており、これらのサブユニットは様々なナトリウムチャネルのアイソフォームと相互作用し、制御しているが、現在までのところ、サブタイプ4のみが不整脈発生と関連している29。 この遺伝子の変異の発生率は調べられていないが、8132>
Jervell-Lange-Nielsen 変異型
この重症型LQTSはIKs電流をコードするKCNQ1、KCNE1遺伝子のホモ接合体30または複合ヘテロ接合体変異によって起こる、つまりLQTS1型やLQTS5型の非常に重症な変異型である。 この疾患は、先天性難聴を伴うのが特徴である。 患者は通常QTc>500msで失神を繰り返し、突然死の危険性が高い。 31
QT延長症候群の診断
Schwartz Score
1985年にSchwartzら32はLQTSの診断基準を発表したが、これは1993年に改訂され、潜在的症例の初期評価に関する重要なガイドラインを含んでいる。 このシステムでは、家族歴、臨床所見、心電図所見に基づき、1~9のスコアを用いる。

QT症候群の出生前診断
胎児の徐脈はLQTSの最初の臨床症状の一つとなりうる。 レトロスペクティブシリーズによると、小児期にLQTSと診断された患者の最大70%が徐脈の病歴を持ち、通常は胎児水腫を伴っている33。 この疾患が強く疑われる場合、妊娠16週以降の羊水穿刺は診断の確定に有用であり、両親のいずれかが特定の変異のキャリアであることが分かっている場合には容易に診断がつく36
LONG QT SYNDROME患者の研究
Clinical History 突然死の家族歴および/または個人歴は、LQTSの診断とリスク層別化のいずれにとっても極めて重要なものである。 さらに、誘発因子や失神の状況により、LQTSの亜型が示されることがある。 疑われる症例の初期評価では、QT 間隔を延長する可能性のある薬物の使用を除外する必要がある
QT 間隔。 QT間隔は、心室再分極の持続時間を示し、Q波の始まりからT波の終わりまでで測定される38。 従来は、Bazett39 が提唱した式を用いて、心拍数に応じて間隔を補正していた(QTc=QT/√RR、単位は秒)。 QT間隔の測定は簡単そうに見えるが、Viskinらによる多施設共同研究40では、心臓専門医以外の医師の40%未満、心臓専門医の50%未満、不整脈専門医の80%以上がQT間隔の正しい測定方法を知っていたという。 自動計測は他の心拍間隔には有効であるが、QT間隔の計算には不正確であるため、医師は手動計測を行い、自動計測を信用しないことが望ましい。 QTは動的な間隔であり、正常範囲はいくつかの要因に依存する。 QTc 間隔が男性で 440 ms、女性で 460 ms の場合、異常とみなされるが、この範囲内には健常者と同様に変異の保因者を見出すことができる(図 3)。 LQTS1の家族において、Vincentら41は遺伝子型が陽性であった症例の中にQTc470msの症例がないことを示した。 Monnigら38は最近、LQTS関連変異のある患者の検出にはQTc>440 msで十分であり、QTc>470 msは症状発現リスクのある患者の特定に有用であり、QTc>500 msは治療中の症状発現患者に認められると発表した
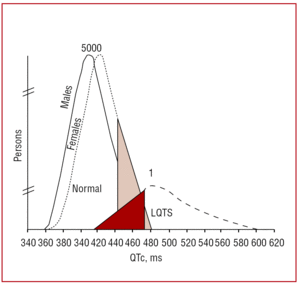
図3.LQTS関連変異を持つ患者 KVLQT1、HERG、またはSCN5Aの変異を持つ患者とその影響を受けていない家族における心拍補正したQT間隔(QTc)の分布を示すモデル。 8132>
QT延長症候群に関連する他の心電図変化
LQTS患者は、極性交替、振幅変動、ノッチ、二相性などの複数のT波の変化を示すことがある42。 T 波オルタナンス(図 4A)は、QRS 群の変動がなく、洞調律 T 波の振幅、形態、極性が 1 拍ごとに変化するものと定義される。 電気的不安定性の指標であり、43 心室再分極の局所的な分散を反映し、時に心室細動に先行する44
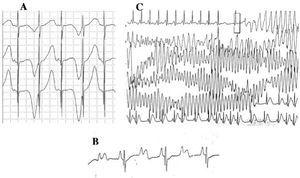
図4. QT延長症候群の心電図変化。 A:T波電気的オルタナンス。 B:房室ブロック2:1。 C:自己限定的なtorsade de pointes。
LQTS患者は洞結節機能障害、徐脈、休止の徴候を伴って進行することがある45。 LQTS1とLQTS3の亜型、特に後者は洞性徐脈を示すことが多く46、一方LQTS4は洞結節機能障害と関連している18
1970-1980年の10年間以来、LQTS47にAV伝導障害が併存することが観察されている(図4B)。 2対1房室ブロックは予後不良のまれな症状であり、持続性徐脈という形で胎児期から存在することがある。 この異常の発生率は4%〜5%と報告されており48、β遮断薬やペースメーカーによる治療にもかかわらず高い死亡率を伴う。49,50 この現象は活動電位の持続時間が長いことによって説明できる。 心室不応期が延長すると、洞房活動の次のインパルスはまだ不応期である心室に到達するためブロックされる。 51 QRS複合体の傾斜は通常急で、ブロックはヒス内領域に限局されている46,51,52 が、ブロックの部位は遺伝子型に依存すると思われる。 現在までにLQTSの2:1ブロックに関連する遺伝子はHERG (LQTS2), 53,54 SCN5A (LQTS3), 52 CACNA1 (LQTS8), 26 and SCN4B (LQTS10) 55
LQTSの特徴的な心室性不整脈はtorsade de pointes (図4C)として知られている. これは、病因に関係なく、QT間隔が延長したときに現れる。 これはリエントリーによる多形性心室頻拍であり、心電図的にはQRS軸が想像上の線を中心に連続的にねじれることで特徴づけられる。 56-58 心室細動や突然死に至ることもある。 これが起こらなければ、患者は失神を経験するだけで、エピソードが短ければ検出されないかもしれない。
ホルター検査は、QT間隔の完全で動的な評価を提供するものである。 時には、洞結節機能障害や房室ブロックのエピソードと同様に、無症候性心室性不整脈の自発的なエピソードが記録されることがある。 59,60 運動負荷試験中の心電図パターンは、LQTS のタイプによって異なる。 LQTS1 の患者は、年齢に応じて計算された最大心拍数に達しないことに加え、頻繁に QT 間隔の増加を示すが、LQTS2 の患者は、予想される心拍数に達しても QT 間隔の増加は軽度か全くない。 ストレス検査は、無症状例における治療効果の評価やリスクの層別化、あるいは不整脈の原因となる事象に疑問がある場合にも有用である。 しかしながら、これらの取り組みから得られた情報は、患者、特に高リスク症例の治療に非常に役立っている。 スクリーニングの主な用途はおそらく遺伝カウンセリングであるが,治療においても重要な意味を持っており,罹患チャンネルに応じて方向性を決めることができる。 ある突然変異の正確な位置は、リスクの進化に関する追加情報を提供することができる。 KCNQ1 の膜貫通領域に変異を有する患者(IKS)は、C 末端領域に変異を有する患者よりも不整脈事象を呈する確率が高い64。同じことが、KCNH2 または HERG65 の孔領域に変異を有する患者にも、N 末端または C 末端領域に変異を有する患者と比較して当てはまる66。
最初のスクリーニングはおそらくKCNQ1、HERG、SCN5A遺伝子に限定され、65%の症例で変異に遭遇する可能性を提供することになる。 8132>
死後の遺伝子スクリーニング
LQTSにつながる遺伝子変異が、突然死を経験した子供や若年成人の不可解な突然死のケースで発見されていることは興味深い。
突然死を経験し、剖検が陰性であった患者の死後の遺伝子研究では、LQTSにつながる変異が様々な割合で示されている67-69:子供では10%近く、若年成人では35%である70-72。 これらの結果に基づいて、すべての新生児に対するルーチンの心電図検査が提案されている。73,74
「分子解剖」としても知られる死後の遺伝子検査は、法的影響に加えて、知らないうちに影響を受けている可能性がある家族にとって重要な意味を持つ。 これらの変化は明らかに病原性ではないが、いくつかは以下のような影響を与える75-78:
1. 不整脈を発症しやすい体質を作る
2.不整脈を発症しやすい体質を作る。 別の非同義変化による病原性の影響を有利にする。
3. 別の非同義変化による病原性の影響を弱める。これはKCNH2(HERG)のK897T多型の場合であり、人口の最大15%に存在して、特定の薬剤に対する感受性と関連しているだけでなく79、同じ遺伝子内の変異の病原性効果を有利にしている78。 もう一つの例は、主に黒人に見られるSCN5A遺伝子のS1103Y多型で、これは13%近い発生率を持ち、小児期の突然死のリスク上昇と関連している80。
興味深いことに、SCN5A遺伝子(ヒトのNav1.5ナトリウムチャネルのアイソフォームをコードする)の産物には、2種類のナトリウムチャネルを生成する2つの代替処理部位が記載されている:一つは1077位(Q1077)のグルタミンを含む2016アミノ酸からなり、もう一つは2015アミノ酸のグルタミンが欠けた(Q1077del)ものだ。 これらの代替処理の転写産物は、同じヒトの心臓に2:1の割合で存在し、いくつかの頻度の高い多型は、文脈がQ1077かQ1077delかによって、チャネル機能に異なる影響を与える。 このことは、最初にSCN5AのH558R多型で示された。SCN5Aは人口の最大30%に存在する。 H558RがQ1077のコンテキストで発現すると、イオン電流の大幅な減少が観察された81。同様の効果がS524Y82多型でも記録されている。 これらの知見は、疾患の重症度の違いや、ある家系で観察される同じ突然変異の異なる表現型を説明する要因となっている。 特に無症状のLQTS1 の検出に有効で、感度92.5%、特異度86%、陽性反応的中度76%、陰性反応的中度96%であった。 また、LQTS2の診断にも有用であるが、感度と特異度は低い。 LQTS3や他の型のLQTSには有用ではない。 正常な状態では、交感神経刺激により IKs カリウムチャネルのリン酸化が誘導され、その機能が最適化され、活動電位の短縮がもたらされる。 LQTS、特に1型の患者では、低用量のアドレナリン(0.025~0.2μg/kg/分)の投与により、QT間隔が30ms83~86以上延長する逆説的反応が観察される
QT 間隔延長と薬剤によるTORSADE DE POINTES
異なる専門分野で用いられる非常に多くの薬剤によってQT間隔の異所的延長を引き起こすことがある。 この望ましくない作用のために市場から撤去された薬剤もある(例えば、アステミゾールやシサプリドなど。詳しくは、www.qtdrugs.org)87,88
非抗不整脈薬による二次性心室性不整脈は、曝露した被験者1万~10万人に1人未満である。 臨床試験の被験者が2000人から3000人であることを考えると、この好ましくない致命的な有害事象は、医薬品開発の臨床段階において容易に発見されなくなるであろう89。 8132>
個人の感受性に関連する要因としては、女性の性別、低カルシウム血症、低マグネシウム血症、徐脈、心不全、心臓手術後、心房細動、左心室肥大、未検出のLQTS、素因の多型、素因の薬剤の高い血清濃度があげられる90。
一般的に薬物と相互作用するチャネルは、その分子構造からKCNH2(HERG)遺伝子にコードされるIKrである。 他のカリウムチャネルは2つのプロリン残基がチャネル孔に向かって角度を変え、チャネル孔の内腔を狭めている。 これに対し、IKrはこれらの残基がないため、より大きな孔前庭が形成され、大きな分子への曝露が容易になる。 さらに、IKrは2つの芳香族残基(チロシンとフェニルアラニン)を持ち、チャネルをブロックするいくつかの薬剤に含まれる芳香族分子との結合を好む。91
上述のように、LQTSの浸透性は不完全であり、無症状の突然変異体保有者がこれらの薬剤を受けることによって悪性の不整脈を起こすことがある。 また、人口的に頻度が高いとされる多型は、ある薬剤を使用した場合に、個々にtorsade de pointesを発症しやすいという特徴を有している。 KCNH2 遺伝子には、健常者において少なくとも 20 種類の多型が報告されており、薬物性不整脈の発症に及ぼす影響についてはまだ解明されていない93。 最大で人口の30%に存在するH558R多型や黒人に頻度の高いS1103Yがこれにあたる80,81,90,94,95。
QT延長症候群と妊娠
LQTSでは遺伝カウンセリングが重要であるが、一般論としてキャリアである女性の妊娠は禁忌ではない、ただし個々のケースは異なるので、適切な状況で個別に評価されるべきである。 一方,特にLQTS2患者では,出産後9か月以内に悪性不整脈を発症しやすいことが報告されている。 このリスクはβ遮断薬治療によりかなり減少する。96
RISK STRATIFICATION
LQTSの進展は様々で、QTc間隔の期間、環境因子、年齢、遺伝子型、治療への反応に影響される97, 98。 心室性不整脈はLQTS1、LQTS2で頻度が高いが、LQTS3ではより重症である。99 前述のように、産後の女性は特に悪性不整脈を起こしやすい。14
QT延長症候群は、以下を伴う場合はハイリスクと考えるべきである。 先天性難聴(Jervell-Lange-Nielsen症候群)
2.先天性難聴(Jervell-Lange-Nielsen症候群)
3. 悪性心室性頻脈性不整脈による再発性失神
3.突然死の家族歴
4.心室性頻脈性不整脈による再発性失神
5.心室性頻脈性不整脈による再発性失神
5.2:1房室ブロック.
6.心筋梗塞.
7.心筋梗塞.
8. T wave electric alternans.
7. LQTS3遺伝子型.
Prioriら97が647人を対象に行った研究では、LQTS1、LQTS2、男性のLQTS3ではQTcが500msだと40歳までに重大イベント(失神、心停止、突然死)を提示する確率が高い(>50%)とされた。 最近,国際的なLQTSレジストリの解析結果が報告された。 2772人の青少年における本疾患の突然死のリスクを解析し,この集団における高リスクと関連する3つの因子が同定された。 QTc>530ms,過去10年間の失神歴,性別である。10~12歳の男子は女子よりリスクが高かったが,13~20歳ではリスクは同等であった。100
TREATMENT
治療を受けていない症候性の患者は,心室性不整脈の初発後に年間死亡率20%,10年死亡率50%と言われている。 症状があるときに治療を確立すべきことは明らかであるが、無症状の患者にどのようなアプローチをとるかはまだ議論中である。 9%の患者では心停止がこの病気の最初の症状であり、48 無症状の患者の12%が症状を発症し、突然死を経験する可能性があることが報告されている。 LQTSの全患者に対して、β遮断薬による初期治療 を開始する必要がある。 運動制限を行うことが望ましいが、臨床的、心電図的なリスクマーカーは意思決定のための有効な基礎となるものである。 上記のように、QT間隔を延長し、心室性不整脈の発生を助長する可能性のあるいくつかの薬剤を使用するリスクについて患者に伝えることが重要である。 遺伝子診断は、この疾患に関する適切な家族カウンセリングを可能にする以外に、予後の評価と具体的な治療の方向付けに役立つものである。 交感神経系によって大きく制御されているIKsチャネル変異(LQTS1)102を持つ患者には特に有効である。 β遮断薬はQT間隔を変化させるのではなく、その分散を変化させる。103 これらの薬剤はイベントの発生率を低下させるが、LQTS1患者の10%、LQTS2患者の23%、LQTS3患者の32%が治療にもかかわらず、心血管症状を有することが示されている106。 特にLQTS3では、重要な効果が得られないようである。実際、LQTS3の心室性不整脈のエピソードは、心拍数が低いときに多く見られるため、これらの患者にはこの薬剤群を注意深く使用することが必要である。 一般論として、β遮断薬治療を開始するまでの5年間に、症状のある患者の32%が症状を再発し、突然死から救出された患者の14%が、この治療のみを受けた場合、5年以内に再び同様の事象を呈すると言われている107。 LQTSの治療には、主にナドロール(0.5-1mg/kg/日)、プロプラノロール(2-4mg/kg/日)、メトプロロール(0.5-1mg/kg/日)、アテノロール(0.5-1mg/kg/日)などのβ遮断薬が使用されている。 しかし、アテノロールはLQTSに有効でない可能性が ある。β遮断薬治療に反応しなかった患者の少なくとも 75%がアテノロールを投与されていたことが報告されているが、この知見は最適ではない用量の 使用に関連している可能性がある104 。 104 適切な投与量を決定するためには、運動負荷試験が有効である。
ナトリウムチャネル遮断薬
LQTS3の原因となるナトリウムチャネル変異は、チャネルの不活性化に不具合をもたらす。 フレカイニドを用いた研究では、心拍数、T波の変化、 QT間隔の改善が報告されている。108 メキシレチンもまた、心電図のリスクマーカーを改善すると報告されている。 8132>
カリウム補給とその利用率を高める薬物
カリウム補給とスピロノラクトンのようなカリウムを節約する薬物は、24%の症例でQTc間隔を短縮させる112,113。 アプリカリム、レブクロマカリム、ニコランジル、ピナシジルなど、カリウムチャネルの開口を促進する薬剤は、LQTSの治療に有用であることが示されている。 114
ペースメーカーと除細動器
ペースメーカー刺激は、休止依存性不整脈の患者に用いられている。115,116 LQTS3患者では徐脈が多いため、通常この治療がより有効である。 DDDペーシングは、一時停止依存性不整脈または高グレードの2:1房室ブロックの患者に適応される。 70拍/分117 未満にプログラムされた周波数は、心室性不整脈の予防には有用でありません。 これらの患者は通常、運動に対して不適切な心拍の加速があるため、センサーを高速応答 にプログラムすることが推奨されます。 ヒステリシスや夜間機能など、休止の存在を示唆する機能はすべて停止しておくこと。 PARP (postventricular atrial refractory period)は可能な限り短くする。 周波数調整機能は、期外収縮後休止を防ぐためにオンにしておく。 T波の過感受性や捕捉の失敗も休止を引き起こす可能性があることを忘れてはならない。 植込み型除細動器(ICD)とβ遮断薬の併用は、突然死の発生率を大幅に減少させる。121 装置のプログラミングは個々の患者のニーズによって異なるが、一般に、無症状で自己限定的なイベントでの治療投与は避けるべきで、そのためには、15秒の検出時間を設定することが望ましい。 AID治療の合併症として、不整脈ストームがある。 この合併症は、ICDのショックに伴う交感神経緊張の亢進が主な原因であり、患者の15%近くが経験する可能性がある118。 1991年、Schwartzら123は、β遮断薬治療の効果が不十分な患者85人の最初のシリーズを発表し、左脚切除術が行われ、5年生存率94%という有望な結果を得た。 現在、この治療法はβ遮断薬治療やペースメーカー植え込みにもかかわらず失神が持続する高リスクの患者や、植え込んだ除細動器から頻繁にショックが起こる患者に提供されている。 この治療法は,単純な左旋毛切断術では十分な効果が得られないため,星状神経節下部と交感神経連鎖のT2~T4左胸部神経節を切除するものである。 微小侵襲的胸腔鏡検査124,125が用いられ、良好な結果を得ている。 この方法で治療した最大の患者シリーズが最近報告され、失神エピソードまたは突然死の数が著しく減少し、5年生存率も95%であることが示された。 失神の既往がある患者の5年生存率は97%で、再発の可能性は11%であり、その大部分は1回の失神で済んだ。 また、左交感神経切除術により、QTセグメントの有意な短縮が認められた。 これらの良好な結果にもかかわらず、突然死の予防は完全ではないが、3%に減少している。 ICDを装着している患者で、除細動器の多重ショックのために手術を受けた場合、平均イベント数は25から0に減少し、95%の減少がみられた。 LQTS1において有益な効果が確認された。 126
アブレーション
心室性不整脈の起始部である期外収縮にアブレーションを行うと、エピソードの発生率が減少することが報告されている127。 しかし、この技術のルーチン的な使用を正当化するための、適切な患者数での長期的な研究はない。
675-82ページの論説を参照
ABBREVIATIONS
AV: atrioventricular
AID: automatic implantable defibrillator
ECG: electrocardiogram
QTc: heart rate-corrected QT
ATS.A
AID:Automatic implantable defibrillatorAID:Atrioventricular。 Andersen-Tawil syndrome
LQTS: QT延長症候群
Medeiros博士はCONACyTとFUNSALUDから経済的支援を受けている。