INLEIDING
Het lange QT-syndroom (LQTS) wordt gekenmerkt door een ernstig veranderde ventriculaire repolarisatie, resulterend in verlenging van het QT-interval op het elektrocardiogram (ECG). De aandoening predisponeert patiënten voor kwaadaardige ventriculaire aritmie (torsade de pointes) en plotse dood. De klinische en elektrocardiografische beschrijving van het lange QT syndroom werd in 1957 gerapporteerd door Anton Jervell en Fred Lange Nielsen,1 die hun studies publiceerden over een familie van niet-consanguine ouders met 6 kinderen. Vier van de kinderen hadden congenitale doofheid en syncopale episoden, en 3 presenteerden plotselinge dood. ECG-onderzoek van deze patiënten toonde een ongewoon lang QT-interval. Beide ouders waren asymptomatisch, hadden een normaal ECG en vertoonden geen gehoorproblemen. In 1964 meldden Romano en Ward onafhankelijk van elkaar een cardiaal syndroom gekenmerkt door recidiverende syncope, een familiegeschiedenis van plotseling overlijden, en verlenging van het QT-interval zonder neuronale doofheid.2 Latere genetische studies toonden aan dat het door Jervell en Lange Nielsen beschreven syndroom, dat gepaard gaat met congenitale neuronale doofheid, overeenkomt met homozygote mutaties, met een ernstig fenotype en een hoog risico op plotseling overlijden. De aandoening die bekend staat als het Romano-Ward-syndroom komt in het algemeen overeen met heterozygote mutaties, de patiënten vertonen geen gehoorveranderingen, en de ernst van de ziekte varieert aanzienlijk. Bijna een halve eeuw later, in 1995,3,4 werden de belangrijkste genen die geassocieerd zijn met LQTS beschreven en werd de ziekte erkend als een cardiale ionkanaalstoornis. Het was de eerste cardiale kanaalpathie die werd beschreven en is tot op heden wellicht de meest uitgebreid onderzochte aritmogene ionkanaalaandoening. Het klinisch beeld varieert sterk: de patiënt kan asymptomatisch zijn, of recidiverende syncope, toevallen of plotselinge dood vertonen als eerste manifestatie van de ziekte. Aanvankelijk werd LQTS beschouwd als een zeldzaam syndroom en in feite is de ernstige presentatie van de ziekte sporadisch. Niettemin wordt de incidentie van verwante mutaties geschat op 1/3000-5000 gevallen,5 32% van de asymptomatische dragers kan een voor de hartslag gecorrigeerd QT-interval (QTc) binnen de normale grenzen hebben, de ziekte wordt overgedragen op 50% van hun nakomelingen, zij zijn gevoeliger voor het ontwikkelen van aritmie in vergelijking met de algemene bevolking, en tot 20% kan symptomatisch worden.6
Het Lange QT syndroom vertoont een grote genetische heterogeniteit. Meer dan 500 mutaties verdeeld over 10 genen zijn bij deze aandoening beschreven: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3, en SCN4B. Ondanks de vooruitgang op dit gebied kan bij 25%-30% van de patiënten geen genetische diagnose worden gesteld.7,8 De presentatie van de ziekte is voornamelijk monogeen6; polygene of samengestelde varianten hebben meestal een ernstiger fenotype. Penetrantie, d.w.z. patiënten die de mutatie hebben en het fenotype manifesteren, varieert van 25% tot 90%.9 Minder vaak kunnen er variaties zijn in de expressiviteit van de ziekte, waarbij verschillende fenotypen het gevolg zijn van dezelfde mutatie. Moleculair genetische studies die de afgelopen 11 jaar zijn ontwikkeld, hebben belangrijke genotype-fenotypecorrelaties opgeleverd, die hebben bijgedragen tot het sturen van de behandelingsaanpak. Bovendien zijn er interessante waarnemingen gedaan over de individuele gevoeligheid voor het ontwikkelen van aritmie in studies waarin de frequente niet-synonieme polymorfismen in deze populatie zijn onderzocht, een aspect dat veel belangstelling heeft gewekt, met name op het gebied van de farmacogenomica.
CLASSIFICATIE VAN HET LANG QT-SYNDROME
Algemene begrippen
De in het verleden gebruikte classificatie van LQTS was gebaseerd op de homozygote of heterozygote presentatie van de ziekte, die aanleiding geeft tot respectievelijk het syndroom van Jervell-Lange-Nielsen (met doofheid) en het syndroom van Romano-Ward (zonder doofheid). De huidige classificatie legt de nadruk op de genetische bevindingen, zoals wordt geïllustreerd in tabel 1. De 3 belangrijkste genen die met de ziekte in verband worden gebracht, werden in 1995-1996 beschreven. Deze genen, die coderen voor de porievormende eenheden van de kaliumkanalen IKs en IKr, en het natriumkanaal Nav1.5, zijn verantwoordelijk voor bijna 65% van de gevallen. Hoewel in de daaropvolgende jaren zeven extra genen in de lijst zijn opgenomen, maken zij slechts 5% van de gevallen uit.
Ion kanalen zijn transmembraan-eiwitten die ionen door het celmembraan transporteren. De bij LQTS betrokken kanalen zijn selectief of gespecialiseerd in het transport van één enkel ion en zijn spanningsafhankelijk, d.w.z. dat hun activering plaatsvindt bij een specifieke intracellulaire spanning, die varieert naargelang het kanaalsubtype. De elektrische en contractiele verschijnselen die in de cardiomyocyt optreden, worden door deze structuren gecontroleerd. Ionenkanalen vormen macromoleculaire complexen bestaande uit een hoofdeenheid die de kanaalporie vormt en hulpeiwitten die deze reguleren (figuur 1). De kanaal disfunctie die in LQTS wordt gezien kan op deze twee plaatsen optreden: het hoofdeiwit of de regulerende eiwitten (Tabel 1). Betrokkenheid van de porievormende eenheid, bekend als alpha, genereert de drie meest voorkomende subtypes van LQTS: LQTS1 (met aantasting van het IKs kalium kanaal), LQTS2 (met aantasting van het IKr kalium kanaal), en LQTS3 (met aantasting van het natrium kanaal). Omdat dit de meest voorkomende subtypes zijn, zijn ze klinisch en genetisch het best gekarakteriseerd. De fenotype-genotype correlaties in deze drie hoofdvormen worden beschreven in figuur 2. Momenteel komt het syndroom van Jervell-Lange-Nielsen overeen met de LQTS 1 en 5 varianten. Kenmerkend is dat deze patiënten congenitale doofheid hebben en samengestelde homozygote of heterozygote mutaties die de IKs-stroom beïnvloeden. Het Romano-Ward syndroom omvat variëteiten van LQTS 1 tot 10 en gaat niet gepaard met doofheid.
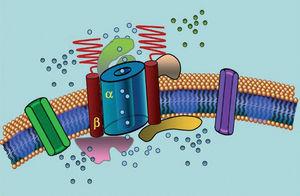
Figuur 1. Schematische voorstelling van het macromoleculaire complex. De ionenkanalen zijn transmembraaneiwitten (α) die door verschillende eiwitten worden gereguleerd; één daarvan is de zogenaamde β-subeenheid.
Lang QT Syndroom Type 1 (LQTS1)
Patiënten met LQTS1 vertonen meestal episoden van ventriculaire aritmie bij inspanning of bij sympathische prikkeling (68%).10 Zwemmen is beschreven als een sport die aritmie triggert bij LQTS1.11 Penetrantie is bijna 62% bij dit subtype. De T-golf bij deze patiënten heeft vaak een brede basis en een zeer verlengde duur12,13 (figuur 2). Het is het meest frequente subtype en verklaart 30%-35% van de gevallen. Het betrokken gen, KvLQT1 (of KCNQ1), bevindt zich op chromosoom 11 (11p15.5) en codeert voor de α-subeenheid van het IKs-kaliumkanaal. De actiepotentiaal wordt verlengd door een vermindering van de uitgaande K+ stroom tijdens fase 3 van de actiepotentiaal.
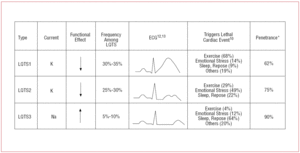
Figuur 2. Genotype-fenotype-correlatie bij de meest voorkomende lange QT-syndromen. *Het betreft gevallen die de mutatie hebben en het fenotype manifesteren.
Lang QT-syndroom type 2 (LQTS2)
Patiënten met LQTS2 hebben de neiging ventriculaire aritmie te vertonen als reactie op emotionele stress (49%) of plotselinge auditieve prikkels (bv. een wekker), en minder vaak tijdens de slaap (22%) of lichaamsbeweging (29%).10 Vooral vrouwen in de postpartumperiode zijn gevoelig.14 De geschatte penetrantie is 79%; daarom kan tot 20% van de gevallen een niet-diagnostisch ECG hebben. De T-golf in LQTS2 is gewoonlijk laag-amplitude en bifid, met notching12,13 (figuur 2). Het betrokken gen is KCNH2 of HERG, gelegen op chromosoom 7 (7q35-36), dat codeert voor het IKr-kaliumkanaal α-subeenheid en verantwoordelijk is voor 25%-30% van de gevallen. Disfunctie van dit kanaal vermindert de uitgaande K+ stroom tijdens fase 3 van de actiepotentiaal, waardoor de duur ervan wordt verlengd.
Lang QT Syndroom Type 3 (LQTS3)
Patiënten met LQTS3 hebben een groter risico op het vertonen van kwaadaardige aritmieën tijdens rust (slaap) of bradycardie.15 Penetrantie van de SCN5A genmutatie is bijna 90%. Het ECG bij LQTS3 vertoont meestal een vertraagde, spitse T-golf en maakt een duidelijke waarneming van de ST-segmentverlenging mogelijk12,13 (figuur 2). Deze patiënten hebben gewoonlijk minder symptomen dan die met LQTS1 of LQTS2, maar de gebeurtenissen zijn karakteristiek dodelijker.
Het aangedane gen in LQTS3 is SCN5A, dat codeert voor de α-subeenheid van het Nav1.5 natriumkanaal (figuur 1), gelegen op chromosoom 3 (3p21-24); het is de oorzaak van de ziekte in 5%-10% van de gevallen. Defecte inactivatie van het kanaal maakt een aanhoudende input van Na+ tijdens fase 2 van de actiepotentiaal mogelijk, waardoor de duur ervan wordt verlengd.
Lang QT Syndroom Type 4 (LQTS4)
Type 4 is een zeldzame variant van LQTS, die bijna 1% van de gevallen uitmaakt. Het is een atypische vorm die een breed spectrum van aritmieën veroorzaakt, waaronder catecholaminerge polymorfe ventriculaire tachycardie, atriumfibrilleren, intraventriculaire geleidingsveranderingen, sinusknoopdisfunctie en bradycardie6-18; bovendien kan de QTc bij veel patiënten binnen de normale grenzen liggen. Het aangetaste gen is ANKB, gelegen op chromosoom 4 (4q25-27), dat codeert voor de synthese van ankyrin-β, een structureel eiwit dat cardiomyocyte membraaneiwitten verbindt met cytoskelet-eiwitten. Deze eiwitten zijn de Na/K ATPase pomp, Na/Ca wisselaar, en inositol trifosfaat receptor (InsP3R). Mutaties die een verlies van ankyrin-β functie veroorzaken, leiden tot een verhoging van de intracellulaire calciumconcentratie en veranderingen in de expressie van N/K ATPase en Na/Ca exchanger. De verhoogde calciumconcentratie geeft aanleiding tot vroege en vertraagde na-depolarisaties. De ventriculaire aritmieën die bij ankyrin-β genmutaties worden waargenomen, zijn dus te wijten aan spontane depolarisaties, gewoonlijk als reactie op catecholaminerge stimulatie.
Lang QT syndroom type 5 (LQTS5)
Type 5 ontstaat door veranderingen in de sequentie van het KCNE1 gen, gelegen op chromosoom 21 (21q22.1p22.)19 KCNE1 codeert voor de synthese van de IKs kanaal β-subeenheid, ook bekend als de minK-subeenheid, die het IKs kanaal reguleert. Dit type maakt minder dan 1% van de gevallen uit.
Lang QT-syndroom type 6 (LQTS6)
Het aangedane gen bij type 6 is KCNE2, gelegen op chromosoom 21 (21q22.1).20 Dit gen codeert voor de kaliumkanaal β-subeenheid, ook bekend als de MiRP1-subeenheid, en het reguleert het IKr-kanaal. Minder dan 1% van de gevallen is van het type 6.
Lang QT-syndroom type 7 of Andersen-Tawil-syndroom (LQTS7)
De dysmorfe bevindingen en elektrocardiografische veranderingen die bij dit syndroom worden gezien, werden voor het eerst beschreven in 1971 door Dr. Andersen21 en in 1994 opnieuw bekeken door Dr. Tawil,22 maar de genetische/moleculaire beschrijving werd pas in 2001 gerapporteerd.23 Nu bekend als het Andersen-Tawil syndroom (ATS) is deze aandoening een autosomaal dominante verandering die wordt gekenmerkt door periodieke verlamming, abnormale ontwikkeling van het skelet, ventriculaire aritmie van het type met frequente ventriculaire extrasystolen, en een bijzondere gevoeligheid voor het ontwikkelen van ventriculair fibrilleren, vooral bij vrouwen. De bij ATS beschreven veranderingen omvatten ventriculaire extrasystolen (41%), niet-onderbroken polymorfe ventriculaire tachycardie (23%), bidirectionele ventriculaire tachycardie (68%) en torsade de pointes (3%).24 Enkele van de waargenomen dysmorfe kenmerken zijn een klein gestalte, scoliose, clinodactylie, hypertelorisme, laag aangezette oren, micrognathie en een breed voorhoofd. De expressie van de ziekte varieert, wat een vroege diagnose bemoeilijkt.23,25 Mutaties in het KCNJ2-gen, gelegen op chromosoom 17 (17q23), dat codeert voor de synthese van het rectificerende kaliumkanaal Kir 2.1, is verantwoordelijk voor 70% van de gevallen. Dit kanaal neemt deel aan fase 4 van de actiepotentiaal. Verscheidene auteurs plaatsen vraagtekens bij de opneming van dit gen in de causale groep van LQTS, omdat het QTc-interval bij dit syndroom slechts licht verlengd is of zelfs normaal, maar de U-golf meestal prominent aanwezig is, hetgeen tot overschatting van het QT-interval heeft geleid. Sommige auteurs suggereren dat KCNJ2-mutaties ATS1 veroorzaken en niet LQTS7.24
Het lange QT-syndroom type 8 (LQTS8)
Type 8 ontstaat door mutaties in het CACNA1-gen, gelegen op chromosoom 12 (12p13.3), dat codeert voor het L-type calciumkanaal Cav1.2. Het veroorzaakt het Timothy-syndroom,26 een aandoening die wordt gekenmerkt door hartafwijkingen, intermitterende immunologische deficiëntie, hypoglykemie, cognitieve veranderingen waaronder autisme, interdigitale vergroeiing, en verlengde QT, wat leidt tot hartritmestoornissen en plotselinge dood.27 Minder dan 0,5% van de gevallen is type 8.
Lang QT-syndroom type 9 (LQTS9)
Deze variant van LQTS ontwikkelt zich door mutaties in het CAV3-gen, gelegen op chromosoom 3 (3p25), dat codeert voor de synthese van caveoline 3. De caveola is een invaginatie van het plasmamembraan die betrokken is bij endocytose, lipidenhomeostase en signaaltransductie. Een belangrijk onderdeel van deze structuur is caveolin, dat 3 bekende subtypes heeft; subtype 3 is specifiek voor skelet- en hartspieren. Sommige ionenkanalen bevinden zich in de caveola, waaronder een cardiale isovorm van het natriumkanaal Nav1.5. Onlangs zijn verschillende mutaties in dit eiwit beschreven. Deze veranderen de biofysische eigenschappen van het natriumkanaal Nav1.5 in vitro, waardoor een fenotype ontstaat dat lijkt op dat van LQTS3.28 Minder dan 1% van de gevallen wordt toegeschreven aan deze oorzaak.
Lang QT-syndroom Type 10 (LQTS10)
Type 10 werd beschreven in een zeer ernstig geval, met QTc >600 ms, foetale bradycardie, en 2:1 atrioventriculair (AV) blok. De ziekte is het gevolg van mutaties in het SCN4B-gen, gelegen op chromosoom 11 (11q23), dat codeert voor de natriumkanaal β4-subeenheid. Er zijn vier verschillende subtypes van de β-subeenheden beschreven, die interageren en de verschillende natriumkanaal-isovormen reguleren; niettemin is tot nu toe alleen subtype 4 in verband gebracht met aritmogenese.29 De incidentie van mutaties van dit gen is niet onderzocht, maar wordt geschat op
Mutaties van de Jervell-Lange-Nielsen variëteit
Deze ernstige vorm van LQTS wordt veroorzaakt door homozygote30 of samengestelde heterozygote mutaties van de KCNQ1, en/of KCNE1 genen, die coderen voor de IKs stroom; d.w.z. een zeer ernstige variëteit van de LQTS1 of LQTS5 vormen. Deze aandoening gaat typisch gepaard met aangeboren doofheid. Patiënten hebben meestal een QTc>500 ms en recidiverende syncope, en lopen een hoog risico op plotseling overlijden. De ouders van patiënten met deze variëteit zijn meestal heterozygoot en hebben een minder ernstige ziekte, of vertonen geen symptomen.31
DIAGNOSIS VAN LONG QT SYNDROME
Schwartz Score
In 1985 publiceerden Schwartz et al32 de criteria voor het stellen van de diagnose LQTS, die in 1993 werden gewijzigd en belangrijke richtlijnen bevatten voor de eerste evaluatie van potentiële gevallen. Dit systeem gebruikt een score van 1 tot 9 gebaseerd op de familiegeschiedenis, en de klinische en electrocardiografische bevindingen. De waarschijnlijkheid van ziekte is laag bij een score van ≥1, gemiddeld bij 2-3, en hoog bij ≥4 (tabel 2).

Prenatale diagnose van het Lange QT Syndroom
Foetale bradycardie kan een van de eerste klinische manifestaties van LQTS zijn. Retrospectieve series hebben aangetoond dat tot 70% van de patiënten bij wie tijdens de kinderjaren LQTS wordt vastgesteld, een voorgeschiedenis van bradycardie hebben, meestal gepaard gaand met foetale hydrops.33 Beoordeling van foetale cardiale repolarisatie tussen week 14 en 39 is nuttig voor een vroege diagnose van LQTS.34
Gonadaal mozaïcisme voor LQTS is in verband gebracht met recidiverende foetale verliezen tijdens het derde trimester van de zwangerschap.35 Bij sterke verdenking op de ziekte kan een vruchtwaterpunctie na 16 weken zwangerschap nuttig zijn voor het stellen van de diagnose, die gemakkelijk te stellen is als van een van de ouders bekend is dat zij drager is van een specifieke mutatie.36
STUDIE VAN EEN PATIËNT MET LONG QT SYNDROME
Klinische voorgeschiedenis
Een familie- en/of persoonlijke voorgeschiedenis van plotseling overlijden is van cruciaal belang voor zowel de diagnose als de risicostratificatie van LQTS. Bovendien kunnen precipiterende factoren en de context van de syncope het LQTS-subtype aangeven. Bij de eerste evaluatie van een verdacht geval moet het gebruik van geneesmiddelen die het QT-interval kunnen verlengen, worden uitgesloten.
QT Interval: Wat is normaal?
Het QT-interval moet bij voorkeur worden gemeten in de afleidingen II of V5,37 waar het een grotere voorspellende waarde blijkt te hebben.38 Dit interval geeft de duur van de ventriculaire repolarisatie aan en wordt gemeten van het begin van de Q-golf tot het einde van de T-golf. Conventioneel wordt de door Bazett39 voorgestelde formule gebruikt om de duur van het interval te corrigeren volgens de hartfrequentie (QTc=QT/√RR, uitgedrukt in seconden). Hoewel de meting van het QT-interval eenvoudig lijkt, wisten in een multicenter studie van Viskin et al,40 minder dan 40% van de artsen die geen cardioloog zijn, minder dan 50% van de cardiologen en meer dan 80% van de specialisten in aritmie hoe ze het interval correct moesten meten. Het is raadzaam dat artsen handmatige metingen uitvoeren en niet vertrouwen op geautomatiseerde metingen, die nuttig kunnen zijn voor andere intervallen, maar onnauwkeurig zijn bij de berekening van het QT-interval. Het QT is een dynamisch interval en de normale grenzen hangen van verschillende factoren af. Hoewel een QTc-interval van é440 ms bij mannen en é460 ms bij vrouwen als abnormaal wordt beschouwd, kan men zowel dragers van mutaties als gezonde personen binnen dit bereik aantreffen (figuur 3). In families met LQTS1 toonden Vincent et al41 aan dat geen van de gevallen met een positief genotype een QTc470 ms had. Monnig et al38 toonden onlangs aan dat QTc>440 ms volstaat om patiënten met LQTS-geassocieerde mutaties op te sporen, QTc>470 ms is nuttig om patiënten met een risico op het ontwikkelen van symptomen te identificeren, en QTc>500 ms wordt aangetroffen bij symptomatische patiënten die een behandeling ondergaan.
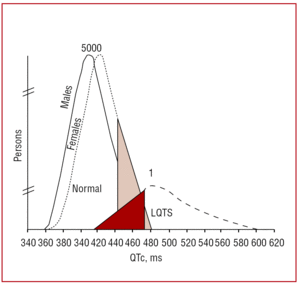
Figuur 3. Model met de verdeling van het hartritmegecorrigeerde QT-interval (QTc) bij patiënten met mutaties in KVLQT1, HERG of SCN5A, en hun niet-aangedane familieleden. De curve links beschrijft de verdeling van de niet getroffen leden en de curve rechts de getroffen leden.
Andere elektrocardiografische veranderingen geassocieerd met het lange QT-syndroom
Patiënten met LQTS kunnen meerdere T-golfveranderingen vertonen: afwisselende polariteit, amplitudevariaties, inkepingen en een bifasisch uiterlijk, onder andere.42 T wave alternans (Figuur 4A) wordt gedefinieerd als een beat-per-beat variatie in amplitude, morfologie en polariteit van een sinusritme T golf, zonder variaties in het QRS complex. Het is een indicator van elektrische instabiliteit,43 die regionale dispersie van ventriculaire repolarisatie weerspiegelt, en gaat soms vooraf aan ventriculaire fibrillatie.44
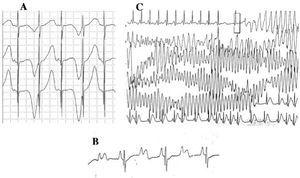
Figuur 4. Elektrocardiografische veranderingen bij het lange QT syndroom. A: T-golf elektrische alternans. B: atrioventriculair blok 2:1. C: zelfbegrensde torsade de pointes.
Patiënten met LQTS kunnen zich ontwikkelen met tekenen van sinusknoop disfunctie, bradycardie, en/of pauzes.45 De subtypes LQTS1 en LQTS3, met name het laatste, vertonen vaak sinusbradycardie,46 terwijl LQTS4 in verband is gebracht met sinusknoopdysfunctie.18
Sinds het decennium 1970-1980 is het naast elkaar bestaan van AV-geleidingsdefecten met LQTS47 waargenomen (figuur 4B). Twee-aan-een AV-blok is een infrequente manifestatie met een slechte prognose die al vanaf de foetale fase aanwezig kan zijn in de vorm van persisterende bradycardie. De incidentie van deze afwijking is gerapporteerd op 4%-5%48 en het is geassocieerd met een hoge mortaliteit ondanks behandeling met bètablokkers en/of pacemakers.49,50 Dit fenomeen kan worden verklaard door een lange duur van de actiepotentiaal. Wanneer de ventriculaire refractaire periode wordt verlengd, wordt de volgende impuls van sinusactiviteit geblokkeerd omdat deze de ventrikels bereikt wanneer deze zich nog in de refractaire periode bevinden. Deze verandering lijkt uitsluitend voor te komen bij LQTS, omdat de ventriculaire refractaire periode groter is dan die van het AV geleidingssysteem.51 De helling van het QRS complex is meestal steil en de blokkade is gelokaliseerd in het infraHis gebied,46,51,52 maar de plaats van de blokkade kan afhankelijk zijn van het genotype. Tot nu toe zijn 4 genen gerelateerd aan 2:1 blok bij LQTS: HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 en SCN4B (LQTS10).55
De karakteristieke ventriculaire aritmie van LQTS staat bekend als torsade de pointes (Figuur 4C). Deze treedt op wanneer het QT-interval verlengd is, ongeacht de etiologie. Het is een polymorfe ventriculaire tachycardie ten gevolge van reentry, die elektrocardiografisch gekenmerkt wordt door een voortdurende verdraaiing van de QRS-as rond een denkbeeldige lijn. Het wordt gewoonlijk voorafgegaan door een pauze gevolgd door een extrasystole (kort-lang-kort RR-interval), zoals in de figuur wordt getoond.56-58 Het kan uitmonden in ventrikelfibrilleren en plotselinge dood. Als dit niet gebeurt, kan de patiënt alleen syncope ervaren, en als de episode kort is, kan deze onopgemerkt blijven.
Holter
Holterstudie biedt een volledige, dynamische beoordeling van het QT-interval. Soms worden spontane episoden van asymptomatische ventriculaire aritmie geregistreerd, evenals episoden van sinusknoopdisfunctie of AV-blok.
Inspanningstest
Patiënten met LQTS kunnen de maximaal verwachte hartfrequentie, berekend op basis van leeftijd, niet bereiken. Bovendien kan het QT-interval onder inspanning paradoxaal gedrag vertonen, door eerder toe te nemen dan af te nemen.59,60 Het elektrocardiografische patroon tijdens inspanningsstresstests zal verschillend zijn, afhankelijk van het type LQTS. Patiënten met LQTS1 bereiken niet alleen niet de voor hun leeftijd berekende maximale hartfrequentie, maar vertonen ook vaak een verhoogd QT-interval, terwijl patiënten met LQTS2 hun verwachte hartfrequentie kunnen bereiken en slechts een lichte of helemaal geen verhoging van het QT-interval vertonen.61,62 In het algemeen hebben patiënten met LQTS3 een fysiologische respons op inspanning, d.w.z. een normale verkorting van het QT-interval.63 Stress-testen kunnen ook nuttig zijn voor het beoordelen van de respons op behandeling en voor het stratificeren van het risico in asymptomatische gevallen, of wanneer er twijfel bestaat over de gebeurtenissen die tot de aritmie leiden.
Genetische screening
In de afgelopen jaren is genetisch onderzoek bij LQTS beperkt gebleven tot onderzoekslaboratoria. Desondanks is de informatie die hieruit is voortgekomen uiterst nuttig geweest voor de behandeling van patiënten, met name in gevallen met een hoog risico. De belangrijkste toepassing van screening is wellicht de genetische counseling, maar zij heeft ook belangrijke implicaties voor de behandeling, die kan worden afgestemd op het getroffen kanaal. De precieze plaats van een bepaalde mutatie kan aanvullende informatie verschaffen over de evolutie van het risico. Patiënten met mutaties in de transmembraanregio van KCNQ1 (IKS) hebben een grotere kans op aritmie dan patiënten met mutaties in de C-terminale regio64 ; hetzelfde geldt voor patiënten met mutaties in de porie-regio van KCNH2 of HERG65 in vergelijking met patiënten met mutaties in de N- of C-terminale regio.66
De initiële screening kan wellicht beperkt blijven tot de KCNQ1, HERG en SCN5A genen, die de mogelijkheid bieden om in 65% van de gevallen mutaties tegen te komen. Wanneer de verkregen resultaten negatief zijn, kan de screening worden uitgebreid tot de genen KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 en SCN4B, waardoor de kans op positieve resultaten met 5% tot 10% toeneemt.
Postmortale genetische screening
Het is interessant dat genmutaties die tot LQTS leiden, zijn gevonden bij kinderen die een plotselinge dood hebben doorgemaakt en in onverklaarbare gevallen van plotselinge dood bij jongvolwassenen.
Postmortale genetische studies van patiënten met plotselinge dood en negatieve autopsie hebben mutaties aangetoond die leiden tot LQTS in variërende percentages67-69: bijna 10% bij kinderen en 35% bij jongvolwassenen.70-72 Op basis van deze resultaten is routinematig ECG-onderzoek bij alle pasgeborenen voorgesteld.73,74
Postmortaal genetisch onderzoek, in de literatuur ook bekend als “moleculaire autopsie”, heeft naast juridische repercussies ook belangrijke implicaties voor families die getroffen kunnen zijn zonder dat zij het weten.
Regelpolymorfismen
Verscheidene frequent voorkomende polymorfismen zijn beschreven in de LQTS-populatie, verdeeld in bijna alle genen die met deze aandoening geassocieerd zijn. Hoewel deze veranderingen blijkbaar niet pathogeen zijn, kunnen sommige de volgende effecten hebben75-78:
1. Veroorzaken van individuele gevoeligheid voor het ontwikkelen van aritmie.
2. 2. Bevorderen van het pathogene effect van een andere niet-synonieme verandering.
3. Verminderen van het pathogene effect van een andere niet-synonieme verandering.
Dit is het geval bij het K897T polymorfisme in KCNH2 (HERG), dat aanwezig is in tot 15% van de bevolking en niet alleen verband houdt met gevoeligheid voor bepaalde geneesmiddelen,79 maar ook het pathogene effect van mutaties in hetzelfde gen bevordert.78 Een ander voorbeeld is het S1103Y polymorfisme in het SCN5A gen, dat vooral bij zwarten voorkomt, een incidentie heeft van bijna 13% en geassocieerd is met een verhoogd risico op plotse dood in de kindertijd.80
Interessant is dat in het product van het SCN5A-gen (dat bij de mens codeert voor de Nav1.5 natriumkanaal-isovorm) twee alternatieve verwerkingsplaatsen zijn beschreven die twee typen natriumkanalen genereren: een met 2016 aminozuren die glutamine bevatten op de 1077-positie (Q1077), en een andere met 2015 aminozuren zonder glutamine (Q1077del). Transcripten van deze alternatieve verwerkingen zijn in een 2:1 verhouding aanwezig in hetzelfde menselijke hart en verschillende frequente polymorfismen zullen verschillende effecten hebben op de werking van het kanaal, afhankelijk van het feit of de context Q1077 of Q1077del is. Dit werd aanvankelijk aangetoond met het H558R polymorfisme van SCN5A, dat aanwezig is in tot 30% van de bevolking. Wanneer H558R werd uitgedrukt in de context van Q1077, werd een sterke vermindering van de ionenstroom waargenomen.81 Een soortgelijk effect werd gedocumenteerd met het S524Y82 polymorfisme. Deze bevindingen hebben factoren opgeleverd die de variërende ernst van de ziekte verklaren, evenals de verschillende fenotypen van dezelfde mutatie die in sommige families zijn waargenomen.77
Farmacologisch testen met adrenaline
Farmacologisch testen met lage-dosis adrenaline is een veilige, nuttige optie om verdachte gevallen van LQTS met een borderline QTc te ontmaskeren. Het is vooral effectief voor het opsporen van asymptomatische vormen van LQTS1, met een sensitiviteit van 92,5%, specificiteit van 86%, positief voorspellende waarde van 76%, en negatief voorspellende waarde van 96%. Het kan ook nuttig zijn bij de diagnose van LQTS2, met lagere sensitiviteit en specificiteit. Het is niet bruikbaar voor LQTS3 of andere vormen van LQTS. Onder normale omstandigheden induceert sympathische stimulatie fosforylering van het IKs kaliumkanaal, waardoor de functie ervan wordt geoptimaliseerd en de actiepotentiaal wordt verkort. Bij patiënten met LQTS, in het bijzonder type 1, wordt een paradoxale reactie op toediening van lage doses adrenaline (0,025-0,2 µg/kg/min) waargenomen die het QT-interval verlengt tot meer dan 30 ms83-86.
QT INTERVAL PROLONGATION AND DRUG-INDUCED TORSADE DE POINTES
Een grote verscheidenheid aan geneesmiddelen die in verschillende medische specialismen worden gebruikt, kan een iatrogene verlenging van het QT-interval veroorzaken. Sommige geneesmiddelen zijn vanwege dit ongewenste effect van de markt gehaald (o.a. astemizol en cisapride; voor meer informatie zie www.qtdrugs.org).87,88
Ventriculaire aritmie secundair aan niet-antiaritmica komt voor bij minder dan één op de 10.000 tot 100.000 blootgestelde personen. Aangezien de klinische studies 2000 tot 3000 proefpersonen omvatten, zou deze ongewenste en fatale bijwerking tijdens de klinische fase van de geneesmiddelenontwikkeling gemakkelijk aan de aandacht ontsnappen89. Dit punt heeft een enorme belangstelling gewekt voor aspecten die betrekking hebben op de veiligheid bij het onderzoek naar en de ontwikkeling van nieuwe geneesmiddelen.
De factoren die verband houden met individuele gevoeligheid zijn onder meer vrouwelijk geslacht, hypocalciëmie, hypomagnesiëmie, bradycardie, hartfalen, postcardioversie, atriumfibrilleren, linkerventrikelhypertrofie, niet-gedetecteerde LQTS, predisponerende polymorfismen, en hoge serumconcentraties van predisponerende geneesmiddelen.90
Het kanaal dat typisch interageert met geneesmiddelen is IKr, gecodeerd door het KCNH2(HERG)-gen, vanwege zijn moleculaire structuur. Andere kaliumkanalen hebben 2 proline residuen die naar de kanaalporie zijn gericht, waardoor het lumen wordt verkleind. In IKr daarentegen ontbreken deze residuen, er ontstaat een grotere porievestibule en de blootstelling aan grote moleculen wordt vergemakkelijkt. Bovendien heeft het 2 aromatische residuen (tyrosine en fenylalanine) die binding bevorderen met aromatische moleculen die aanwezig zijn in verschillende geneesmiddelen die het kanaal kunnen blokkeren.91
Zoals hierboven vermeld, is de penetrantie van LQTS onvolledig en sommige asymptomatische dragers van mutaties kunnen kwaadaardige aritmie vertonen na toediening van een van deze geneesmiddelen. Bovendien geven polymorfismen die als frequent in de bevolking worden beschouwd, individuele gevoeligheid voor de ontwikkeling van torsade de pointes bij gebruik van bepaalde geneesmiddelen. Dit is het geval voor het R1047L polymorfisme, het op één na meest frequente in KCNH2, dat in verband werd gebracht met de ontwikkeling van torsade de pointes bij gebruik van het geneesmiddel dofetilide.92 Er zijn ten minste 20 KCNH2 genpolymorfismen beschreven bij gezonde personen en hun effect op de individuele gevoeligheid voor het ontwikkelen van geneesmiddelgerelateerde aritmie moet nog worden bepaald.93 Polymorfismen die gevoeligheid verlenen voor het ontwikkelen van ventriculaire aritmie zijn ook gedocumenteerd in het natriumkanaal Na1.5. Dit is het geval voor het H55 genpolymorfisme, dat in de bevolking als frequent wordt beschouwd. Dit is het geval voor het H558R polymorfisme, dat bij tot 30% van de bevolking voorkomt, of voor S1103Y, dat frequent voorkomt bij zwarten80,81,90,94,95; Hun implicatie in drug-induced susceptibility is niet onderzocht.
LONG QT SYNDROME EN PREGNANCY
Genetische counseling is belangrijk bij LQTS, maar in het algemeen is er geen contra-indicatie voor zwangerschap bij vrouwen die draagster zijn, hoewel elk geval anders is en individueel moet worden beoordeeld in de juiste context.
Er is opgemerkt dat het risico op het presenteren van maligne ventriculaire aritmie afneemt met de zwangerschap. Daarentegen is een grotere kwetsbaarheid voor het presenteren van maligne aritmie gerapporteerd binnen de eerste 9 maanden na de bevalling, met name bij patiënten met LQTS2. Dit risico neemt aanzienlijk af met therapie met bètablokkers.96
RISICO STRATIFICATIE
De evolutie van LQTS varieert en wordt beïnvloed door de duur van het QTc-interval, omgevingsfactoren, leeftijd, genotype, en respons op behandeling.97,98 Ventriculaire aritmie komt vaker voor bij LQTS1 en LQTS2, maar is ernstiger bij LQTS3.99 Zoals gezegd zijn vrouwen vooral vatbaar voor maligne aritmie tijdens de postpartumperiode.14
Het lange QT-syndroom moet als risicovol worden beschouwd wanneer het gepaard gaat met het volgende:
1. Aangeboren doofheid (syndroom van Jervell-Lange-Nielsen).
2. Recidiverende syncope als gevolg van kwaadaardige ventriculaire tachyaritmie.
3. Familieanamnese van plotseling overlijden.
4. QTc>500 ms.
5. 2:1 atrioventriculair blok.
6. T wave electric alternans.
7. LQTS3 genotype.
Uit de studie van Priori et al97, uitgevoerd bij 647 patiënten, bleek dat de kans op het optreden van een belangrijke gebeurtenis (syncope, hartstilstand, plotseling overlijden) vóór de leeftijd van 40 jaar groot is (>50%) wanneer de QTc >500 ms is bij LQTS1, LQTS2, en bij mannen met LQTS3. Onlangs werd een analyse van het internationale LQTS-register gerapporteerd. Het risico op plotse dood werd geanalyseerd in 2772 adolescenten met de ziekte, en 3 factoren geassocieerd met een hoger risico in deze populatie werden geïdentificeerd: QTc>530 ms, voorgeschiedenis van syncope in de afgelopen 10 jaar, en geslacht; 10- tot 12-jarige jongens hadden een hoger risico dan meisjes, maar in de leeftijdsgroep van 13 tot 20 jaar was het risico vergelijkbaar.100
TREATMENT
Symptomatische patiënten die geen behandeling krijgen, hebben een jaarlijks sterftecijfer van 20% en een 10-jaars sterftecijfer van 50% na een eerste voorval van ventriculaire aritmie. Hoewel het duidelijk is dat behandeling moet worden ingesteld wanneer er symptomen zijn, wordt nog steeds gediscussieerd over de te volgen aanpak bij asymptomatische patiënten. Er is gedocumenteerd dat hartstilstand bij 9% van de patiënten de eerste manifestatie van de ziekte kan zijn,48 en dat 12% van de asymptomatische patiënten symptomen zal ontwikkelen en plotseling kan overlijden. Bij alle patiënten met LQTS moet een initiële behandeling met bètablokkers worden gestart. Beperking van de inspanning is aan te bevelen, maar de klinische en elektrocardiografische risicomarkers zijn een nuttige basis voor de besluitvorming. Het is belangrijk patiënten te informeren over het risico van het gebruik van verschillende geneesmiddelen die het QT-interval kunnen verlengen en de ontwikkeling van ventriculaire aritmie kunnen bevorderen, zoals hierboven is vermeld. Genetische diagnose maakt niet alleen een passende gezinsbegeleiding in verband met de ziekte mogelijk, maar is ook een hulp bij het beoordelen van de prognose en het oriënteren van een specifieke behandeling.
Bètablokkers
Bètablokkers zijn de eerstelijnsbehandeling voor LQTS en alle patiënten moeten ze als eerste therapie krijgen.101 Zij geven een vermindering van het risico op cardiovasculaire gebeurtenissen tot 64%100 en zijn bijzonder effectief bij patiënten met IKs kanaalmutaties (LQTS1),102 die in hoge mate worden gereguleerd door het sympatische systeem. Bètablokkers veranderen het QT-interval niet, maar wel de dispersie ervan.103 Hoewel deze geneesmiddelen de incidentie van voorvallen verminderen,104,105 is aangetoond dat 10% van de patiënten met LQTS1, 23% met LQTS2 en 32% met LQTS3 cardiovasculaire symptomen zullen hebben ondanks behandeling.106 Met name patiënten met LQTS3 lijken geen belangrijke voordelen te behalen; in feite moet deze groep geneesmiddelen bij deze patiënten met voorzichtigheid worden gebruikt, omdat episoden van ventriculaire aritmie bij LQTS3 vaker voorkomen wanneer de hartfrequentie laag is. In het algemeen zal 32% van de symptomatische patiënten in de eerste 5 jaar voor het begin van de behandeling met bètablokkers recidiverende symptomen vertonen, en zal 14% van de patiënten die van een plotselinge doodsepisode zijn gered binnen 5 jaar opnieuw een soortgelijke gebeurtenis vertonen als zij alleen deze therapie krijgen.107 Bij de behandeling van LQTS zijn verschillende bètablokkers gebruikt, voornamelijk nadolol (0,5-1 mg/kg/dag), propranolol (2-4 mg/kg/dag), metoprolol (0,5-1 mg/kg/dag), en atenolol (0,5-1 mg/kg/dag). Het is echter mogelijk dat atenolol niet gunstig is bij LQTS; er is gemeld dat ten minste 75% van de patiënten die niet reageerden op bètablokkertherapie atenolol kregen, hoewel deze bevinding verband kan houden met het gebruik van suboptimale doses.104 Oefentests zijn nuttig om de juiste dosis vast te stellen. De maximale hartfrequentie mag tijdens de behandeling niet hoger zijn dan 130 slagen/min.
Natriumkanaalblokkers
De natriumkanaalmutaties die LQTS3 veroorzaken, veroorzaken een defecte inactivering van het kanaal; natriumkanaalblokkering is nuttig gebleken bij deze patiënten. Studies met flecainide hebben verbeteringen aangetoond in de hartfrequentie, T-golf veranderingen en QT-interval.108 Mexiletine zou ook de elektrocardiografische risicomarkers verbeteren.63,109,110 In vitro studies met ranolazine hebben verminderingen aangetoond van de schadelijke effecten van mutaties bij mensen.111 Hoewel de resultaten bemoedigend zijn, moet men in gedachten houden dat er geen lange-termijn studies zijn die deze therapie evalueren, en geen gerapporteerde bevindingen van grote series. Natriumkanaalblokkers mogen niet worden toegediend als er geen bevestigde genetische diagnose is.
Kaliumsuppletie en geneesmiddelen die de beschikbaarheid ervan vergroten
Kaliumsupplementen en/of kaliumsparende geneesmiddelen, zoals spironolacton, verkorten het QTc-interval in 24% van de gevallen.112,113 Geneesmiddelen die het openen van de kaliumkanalen bevorderen, zoals aprikalim, levcromakalim, nicorandil, en pinacidil, zijn nuttig gebleken bij de behandeling van LQTS. De subtypen waarbij zij van bijzonder nut zijn, zijn LQTS1 en LQTS2.114
Pacemakers en defibrillatoren
Pacemakerstimulatie is toegepast bij patiënten met pauze-afhankelijke aritmie.115,116 Patiënten met LQTS3 hebben meestal meer baat bij deze behandeling omdat de prevalentie van bradycardie in deze groep groter is. DDD-pacing is geïndiceerd bij patiënten met pauze-afhankelijke aritmie of een 2:1 AV-blok van hoge graad. Frequenties geprogrammeerd onder 70 slagen/min117 zijn niet nuttig voor het voorkomen van ventriculaire aritmie. Het wordt aanbevolen de sensor op snelle respons te programmeren, omdat deze patiënten gewoonlijk een ongepaste hartslagversnelling vertonen als reactie op inspanning. Alle functies die de aanwezigheid van pauzes impliceren, zoals de hysterese en de nachtfunctie, moeten worden uitgeschakeld. De PARP (postventriculaire atriale refractaire periode) moet zo kort mogelijk zijn. De frequentieregelfunctie moet aan staan om een postextrasystolische pauze te voorkomen. Er zij aan herinnerd dat T-golf oversensing en capture failures ook aanleiding kunnen geven tot pauzes. Gecombineerd gebruik van een implanteerbare cardioverter defibrillator (ICD) en bètablokkers vermindert de incidentie van plotse dood aanzienlijk.118-120 De indicatie voor deze maatregelen is duidelijk in gevallen met een hoog risico.121 De programmering van het apparaat zal variëren naar gelang van de behoeften van de individuele patiënt, maar over het algemeen moet toediening van behandeling bij asymptomatische, zelfbegrensde voorvallen worden vermeden; hiertoe is een detectietijd van 15 s geïndiceerd. Aritmestorm is een complicatie van AID-therapie. Bij bijna 15% van de patiënten kan deze complicatie optreden, die voor een groot deel te wijten is aan een verhoogde sympathische tonus na de ICD-schok.118 Dit probleem kan worden beheerst door de dosis bètablokkers te verhogen. Indien deze maatregel geen nut heeft, moet resectie van de sympathische ketenganglia worden overwogen.
Linker-symfathectomie
In 1971 werd sympathische gangliectomie geïntroduceerd als een nuttige therapeutische optie bij deze patiënten.122 In 1991 publiceerden Schwartz et al123 de eerste serie van 85 patiënten met een slechte respons op behandeling met bètablokkers, bij wie een linker stellectomie werd uitgevoerd met bemoedigende resultaten: een 5-jaars overlevingspercentage van 94%. Momenteel wordt deze therapeutische optie aangeboden aan patiënten met een hoog risico die syncope blijven houden ondanks behandeling met bètablokkers en/of implantatie van een pacemaker, en aan patiënten die frequent schokken van hun geïmplanteerde defibrillator ondervinden. De procedure bestaat uit resectie van het inferieure deel van het ganglion stellate en de T2 tot T4 linker thoracale ganglia van de sympathische keten, aangezien eenvoudige linker stellectomie niet voldoende doeltreffend is gebleken. Micro-invasieve thoracoscopie124,125 is gebruikt met goede resultaten. De grootste serie patiënten die met deze methode werd behandeld, werd onlangs gerapporteerd en toonde een significante vermindering van het aantal syncope-episoden of plotselinge sterfgevallen, alsmede een 5-jaars overlevingskans van 95%. Bij patiënten met eerdere syncope was de 5-jaarsoverleving 97%, met een recidiefkans van 11%, die in de meerderheid bestond uit een enkele syncope-episode. Er was ook een significante vermindering van het QT segment na linker sympathectomie. Ondanks deze gunstige resultaten is de preventie van plotse dood niet volledig, maar wel teruggebracht tot 3%. Bij patiënten met een ICD die een operatie ondergingen vanwege meerdere defibrillatorschokken, daalde het gemiddelde aantal voorvallen van 25 tot 0, een reductie van 95%. Een gunstig effect werd bevestigd bij LQTS1. De voordelen zijn waarschijnlijk kleiner bij patiënten met LQTS2, en bij LQTS3 is de effectiviteit niet bewezen.126
Ablatie
Er is gerapporteerd dat ablatie van de extrasystole, die in sommige gevallen de ventriculaire aritmie initieert, kan worden uitgevoerd met een vermindering van de incidentie van episodes.127 Er zijn echter geen langetermijnstudies met een voldoende aantal patiënten om routinematig gebruik van deze techniek te rechtvaardigen.
Zie het redactioneel op blz. 675-82
ABBREVIATIES
AV: atrioventriculair
AID: automatische implanteerbare defibrillator
ECG: elektrocardiogram
QTc: hartritmegecorrigeerd QT
ATS: Andersen-Tawil-syndroom
LQTS: lang QT-syndroom
Dr. Medeiros ontvangt economische steun van CONACyT en FUNSALUD.