WPROWADZENIE
Zespół długiego QT (LQTS) charakteryzuje się silnie zmienioną repolaryzacją komór, co skutkuje wydłużeniem odstępu QT w elektrokardiogramie (EKG). Stan ten predysponuje pacjentów do złośliwych komorowych zaburzeń rytmu serca (torsade de pointes) i nagłego zgonu. Opis kliniczny i elektrokardiograficzny zespołu długiego QT został przedstawiony w 1957 roku przez Antona Jervella i Freda Lange Nielsena,1 którzy opublikowali wyniki swoich badań na temat rodziny rodziców niebędących krewnymi, w której było 6 dzieci. Czworo z nich miało wrodzoną głuchotę i epizody synkopalne, a troje prezentowało nagły zgon. Badanie EKG u tych pacjentów wykazało nietypowo długi odstęp QT. Oboje rodzice byli bezobjawowi, mieli prawidłowe EKG i nie wykazywali problemów ze słuchem. W 1964 roku Romano i Ward niezależnie od siebie opisali zespół kardiologiczny charakteryzujący się nawracającymi omdleniami, rodzinnym występowaniem nagłej śmierci i wydłużeniem odstępu QT bez głuchoty neuronalnej.2 Późniejsze badania genetyczne wykazały, że zespół opisany przez Jervella i Lange Nielsena, któremu towarzyszy wrodzona głuchota neuronalna, odpowiada homozygotycznym mutacjom, z ciężkim fenotypem i dużym ryzykiem nagłej śmierci. Stan znany jako zespół Romano-Warda odpowiada zazwyczaj mutacjom heterozygotycznym, pacjenci nie wykazują zmian słuchowych, a ciężkość choroby jest bardzo zróżnicowana. Prawie pół wieku później, w 1995 roku,3,4 opisano główne geny związane z LQTS i uznano chorobę za zaburzenie kanałów jonowych serca. Była to pierwsza opisana kardiochirurgiczna kanałopatia i jest prawdopodobnie najszerzej badanym zaburzeniem arytmogennych kanałów jonowych. Obraz kliniczny jest bardzo zróżnicowany: pacjent może być bezobjawowy lub wykazywać nawracające omdlenia, napady drgawkowe lub nagły zgon jako pierwszą manifestację choroby. Początkowo LQTS uważany był za zespół rzadki i w efekcie ciężka postać choroby występuje sporadycznie. Mimo to częstość występowania związanych z nim mutacji szacuje się na 1/3000-5000 przypadków,5 32% bezobjawowych nosicieli może mieć skorygowany odstęp QT (QTc) w granicach normy, choroba jest przekazywana 50% ich potomków, są oni bardziej podatni na rozwój arytmii w porównaniu z populacją ogólną, a do 20% może dawać objawy.6
Zespół długiego QT wykazuje dużą heterogenność genetyczną. W schorzeniu tym opisano ponad 500 mutacji rozmieszczonych w 10 genach: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 i SCN4B. Pomimo postępu w tej dziedzinie, nie udaje się ustalić rozpoznania genetycznego u 25%-30% pacjentów.7,8 Obraz choroby jest głównie monogenowy6; odmiany poligenowe lub złożone mają zwykle cięższy fenotyp. Penetrancja, czyli pacjenci, którzy mają mutację i przejawiają fenotyp, waha się od 25% do 90%.9 Rzadziej mogą występować różnice w ekspresyjności choroby, z kilkoma fenotypami wynikającymi z tej samej mutacji. Badania z zakresu genetyki molekularnej prowadzone w ciągu ostatnich 11 lat pozwoliły uzyskać istotne korelacje genotyp-fenotyp, które pomogły ukierunkować podejście do leczenia. Ponadto w badaniach nad częstymi w tej populacji polimorfizmami nonsynonimicznymi poczyniono interesujące obserwacje dotyczące indywidualnej podatności na wystąpienie arytmii, co wzbudziło duże zainteresowanie, szczególnie w dziedzinie farmakogenomiki.
KLASYFIKACJA SYNDROMU DŁUGIEGO QT
Pojęcia ogólne
Klasyfikacja LQTS stosowana w przeszłości opierała się na homozygotycznej lub heterozygotycznej prezentacji choroby, co daje początek odpowiednio zespołowi Jervella-Lange-Nielsena (z głuchotą) i zespołowi Romano-Warda (bez głuchoty). Obecna klasyfikacja kładzie nacisk na wyniki badań genetycznych, co ilustruje tabela 1. 3 główne geny związane z chorobą zostały opisane w latach 1995-1996. Geny te, kodujące jednostki tworzące pory kanałów potasowych IKs i IKr oraz kanału sodowego Nav1.5, odpowiadają za blisko 65% przypadków. Chociaż w kolejnych latach do listy dołączono siedem dodatkowych genów, odpowiadają one jedynie za 5% przypadków.
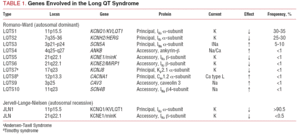
Kanały jonowe są białkami transmembranowymi, które transportują jony przez błonę komórkową. Kanały, których dotyczy LQTS są selektywne lub wyspecjalizowane w transporcie jednego jonu i są zależne od napięcia, tzn. ich aktywacja następuje przy określonym napięciu wewnątrzkomórkowym, które różni się w zależności od podtypu kanału. Zjawiska elektryczne i kurczliwe zachodzące w kardiomiocycie są kontrolowane przez te struktury. Kanały jonowe tworzą wielkocząsteczkowe kompleksy składające się z jednostki głównej, która tworzy por kanału oraz białek pomocniczych, które go regulują (ryc. 1). Zaburzenia funkcji kanałów obserwowane w LQTS mogą występować w tych dwóch miejscach: w białku głównym lub w białkach regulujących (tab. 1). Zaangażowanie jednostki tworzącej pory, znanej jako alfa, generuje trzy najczęstsze podtypy LQTS: LQTS1 (wpływający na kanał potasowy IKs), LQTS2 (wpływający na kanał potasowy IKr) i LQTS3 (wpływający na kanał sodowy). Ponieważ są to najczęstsze podtypy, są one najlepiej scharakteryzowane klinicznie i genetycznie. Korelacje fenotyp-genotyp w tych trzech głównych postaciach opisano na rycinie 2. Obecnie zespół Jervella-Lange-Nielsena odpowiada odmianom LQTS 1 i 5. Charakterystyczna dla tych pacjentów jest wrodzona głuchota i złożone homozygotyczne lub heterozygotyczne mutacje, które wpływają na prąd IKs. Zespół Romano-Warda obejmuje odmiany od LQTS 1 do 10 i nie wiąże się z głuchotą.
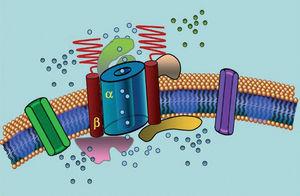
Rysunek 1. Schematyczne przedstawienie kompleksu wielkocząsteczkowego. Kanały jonowe są białkami transmembranowymi (α) regulowanymi przez różne białka; jednym z nich jest tzw. podjednostka β.
Zespół długiego QT typu 1 (LQTS1)
Pacjenci z LQTS1 zwykle prezentują epizody arytmii komorowej podczas wysiłku fizycznego lub pod wpływem bodźców współczulnych (68%).10 Pływanie zostało opisane jako sport wyzwalający arytmię w LQTS1.11 Penetralność wynosi prawie 62% w tym podtypie. Załamek T u tych pacjentów często ma szeroką podstawę i bardzo wydłużony czas trwania12,13 (ryc. 2). Jest to najczęstszy podtyp i wyjaśnia 30%-35% przypadków. Dotknięty gen KvLQT1 (lub KCNQ1) jest zlokalizowany na chromosomie 11 (11p15.5) i koduje podjednostkę α kanału potasowego IKs. Wydłużenie potencjału czynnościowego jest spowodowane zmniejszeniem prądu K+ wychodzącego podczas fazy 3 potencjału czynnościowego.
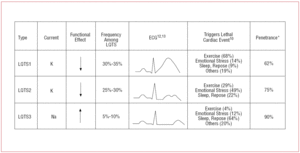
Ryc. 2. Korelacja genotyp-fenotyp w najczęstszych zespołach długiego QT. *Odnosi się do przypadków, które mają mutację i manifestują fenotyp.
Zespół długiego QT typu 2 (LQTS2)
Pacjenci z LQTS2 mają tendencję do występowania komorowych zaburzeń rytmu w odpowiedzi na stres emocjonalny (49%) lub nagłe bodźce słuchowe (np. budzik), rzadziej podczas snu (22%) lub wysiłku fizycznego (29%).10 Szczególnie podatne są kobiety w okresie poporodowym.14 Szacuje się, że penetracja wynosi 79%, dlatego nawet 20% przypadków może mieć niediagnostyczny zapis EKG. Załamek T w LQTS2 jest zwykle niskoamplitudowy i dwudzielny, z wcięciem12,13 (ryc. 2). Uszkodzonym genem jest KCNH2 lub HERG, zlokalizowany na chromosomie 7 (7q35-36), który koduje podjednostkę α kanału potasowego IKr i odpowiada za 25%-30% przypadków. Dysfunkcja tego kanału zmniejsza natężenie wychodzącego prądu K+ podczas 3. fazy potencjału czynnościowego, wydłużając jego czas trwania.
Zespół długiego QT typu 3 (LQTS3)
Pacjenci z LQTS3 mają większe ryzyko wystąpienia złośliwych arytmii podczas spoczynku (snu) lub bradykardii.15 Penetrancja mutacji genu SCN5A wynosi blisko 90%. EKG w LQTS3 zwykle wykazuje opóźnioną, spiczastą falę T i pozwala na wyraźną obserwację wydłużenia odcinka ST12,13 (ryc. 2). Pacjenci ci mają zwykle mniej objawów niż pacjenci z LQTS1 lub LQTS2, ale zdarzenia są charakterystycznie bardziej śmiertelne.
Dotkniętym genem w LQTS3 jest SCN5A, który koduje podjednostkę α kanału sodowego Nav1.5 (ryc. 1), zlokalizowany na chromosomie 3 (3p21-24); jest on przyczyną choroby w 5%-10% przypadków. Wadliwa inaktywacja kanału umożliwia utrzymywanie się napływu Na+ w fazie 2 potencjału czynnościowego, wydłużając czas jego trwania.
Zespół długiego QT typu 4 (LQTS4)
Typ 4 jest rzadką odmianą LQTS, stanowiącą blisko 1% przypadków. Jest to postać atypowa, która powoduje szerokie spektrum arytmii, w tym katecholaminergiczny polimorficzny częstoskurcz komorowy, migotanie przedsionków, zmiany przewodzenia śródkomorowego, dysfunkcję węzła zatokowego i bradykardię6-18; ponadto u wielu pacjentów QTc może mieścić się w granicach normy. Genem, którego dotyczy choroba, jest ANKB, zlokalizowany na chromosomie 4 (4q25-27), który koduje syntezę ankiryny-β, białka strukturalnego łączącego białka błonowe kardiomiocytów z białkami cytoszkieletu. Białka te to pompa Na/K ATPazy, wymiennik Na/Ca oraz receptor dla trójfosforanu inozytolu (InsP3R). Mutacje powodujące utratę funkcji ankiryny-β prowadzą do wzrostu wewnątrzkomórkowego stężenia wapnia oraz zmiany ekspresji pompy N/K ATPaza i wymiennika Na/Ca. Podwyższone stężenie wapnia prowadzi do wczesnej i opóźnionej depolaryzacji następczej. Tak więc arytmie komorowe obserwowane w mutacjach genu ankiryny-β są wynikiem spontanicznych depolaryzacji, zwykle w odpowiedzi na stymulację katecholaminergiczną.
Zespół długiego QT typu 5 (LQTS5)
Typ 5 powstaje w wyniku zmian w sekwencji genu KCNE1 zlokalizowanego na chromosomie 21 (21q22.1p22.)19 KCNE1 koduje syntezę podjednostki β kanału IKs, zwanej również podjednostką minK, która reguluje pracę kanału IKs. Ten typ stanowi mniej niż 1% przypadków.
Zespół długiego QT typu 6 (LQTS6)
Aktualnym genem w typie 6 jest KCNE2, zlokalizowany na chromosomie 21 (21q22.1).20 Gen ten koduje podjednostkę β kanału potasowego, znaną również jako podjednostka MiRP1, i reguluje kanał IKr. Mniej niż 1% przypadków to zespół typu 6.
Zespół długiego QT typu 7 lub zespół Andersena-Tawila (LQTS7)
Cechy dysmorficzne i zmiany elektrokardiograficzne obserwowane w tym zespole zostały po raz pierwszy opisane w 1971 roku przez dr Andersena21 i ponownie opisane w 1994 roku przez dr Tawila,22 ale opis genetyczny/molekularny został przedstawiony dopiero w 2001 roku.23 Znany obecnie jako zespół Andersena i Tawila (ATS) zespół ten jest autosomalną dominującą zmianą charakteryzującą się okresowym porażeniem, nieprawidłowym rozwojem kośćca, komorowymi zaburzeniami rytmu o typie częstych ekstrasystolii komorowych oraz szczególną podatnością na wystąpienie migotania komór, zwłaszcza u kobiet. Zmiany opisywane w ATS obejmują ekstrasystole komorowe (41%), nieutrwalony polimorficzny częstoskurcz komorowy (23%), dwukierunkowy częstoskurcz komorowy (68%) i torsade de pointes (3%).24 Niektóre z obserwowanych cech dysmorficznych to: niski wzrost, skolioza, klinodaktylia, hiperteloryzm, niskie osadzenie uszu, mikrognacja i szerokie czoło. Ekspresja choroby jest różna, co utrudnia wczesną diagnostykę.23,25 Mutacje w genie KCNJ2 zlokalizowanym w chromosomie 17 (17q23), który koduje syntezę prostowniczego kanału potasowego Kir 2.1, odpowiadają za 70% przypadków. Kanał ten bierze udział w fazie 4 potencjału czynnościowego. Kilku autorów kwestionuje włączenie tego genu do grupy przyczynowej LQTS, ponieważ odstęp QTc jest w tym zespole tylko nieznacznie wydłużony lub nawet prawidłowy, natomiast załamek U jest zwykle wybitny, co prowadziło do zawyżania wartości odstępu QT. Czytelnik zauważy, że niektórzy autorzy sugerują, iż mutacje KCNJ2 generują ATS1, a nie LQTS7.24
Zespół długiego QT typu 8 (LQTS8)
Typ 8 powstaje w wyniku mutacji w genie CACNA1 zlokalizowanym na chromosomie 12 (12p13.3), który koduje kanał wapniowy typu L Cav1. 2.Powoduje on zespół Timothy’ego26 , charakteryzujący się wadami serca, okresowym niedoborem odporności, hipoglikemią, zaburzeniami poznawczymi, w tym autyzmem, fuzją międzypalcową oraz wydłużeniem odcinka QT, co prowadzi do zaburzeń rytmu serca i nagłej śmierci27. Mniej niż 0,5% przypadków to typ 8.
Zespół długiego QT typu 9 (LQTS9)
Ta odmiana LQTS rozwija się w wyniku mutacji w genie CAV3, zlokalizowanym na chromosomie 3 (3p25), który koduje syntezę kaweoliny 3. Jaskinia (caveola) jest wgłębieniem błony plazmatycznej biorącym udział w endocytozie, homeostazie lipidów i transdukcji sygnału. Ważnym składnikiem tej struktury jest kaweolina, która ma 3 znane podtypy; podtyp 3 jest specyficzny dla mięśni szkieletowych i mięśnia sercowego. Niektóre kanały jonowe są kolokalizowane w jaskini, w tym sercowa izoforma kanału sodowego Nav1.5. Ostatnio opisano kilka mutacji tego białka. Zmieniają one właściwości biofizyczne kanału sodowego Nav1.5 in vitro, generując fenotyp podobny do obserwowanego w LQTS3.28 Mniej niż 1% przypadków przypisuje się tej przyczynie.
Zespół długiego QT typu 10 (LQTS10)
Typ 10 opisano w bardzo ciężkim przypadku, z QTc >600 ms, bradykardią płodową i blokiem przedsionkowo-komorowym (AV) 2:1. Wynika ona z mutacji w genie SCN4B, zlokalizowanym na chromosomie 11 (11q23), który koduje podjednostkę β4 kanału sodowego. Opisano cztery różne podtypy podjednostek β, które oddziałują ze sobą i regulują różne izoformy kanału sodowego, jednak dotychczas tylko podtyp 4 był związany z arytmogenezą.29 Częstość występowania mutacji tego genu nie została zbadana, ale szacuje się ją na
Mutacje odmiany Jervell-Lange-Nielsen
Ta ciężka postać LQTS jest spowodowana homozygotycznymi30 lub złożonymi heterozygotycznymi mutacjami genów KCNQ1 i/lub KCNE1, które kodują prąd IKs; czyli jest to bardzo ciężka odmiana postaci LQTS1 lub LQTS5. Choroba ta jest charakterystycznie związana z wrodzoną głuchotą. Pacjenci mają zwykle QTc>500 ms, nawracające omdlenia i są w grupie wysokiego ryzyka nagłego zgonu. Rodzice pacjentów z tą odmianą są zwykle heterozygotyczni i mają mniej nasiloną chorobę lub nie wykazują żadnych objawów.31
DIAGNOZA SYNDROMU DŁUGIEGO QT
Szwartz Score
W 1985 roku Schwartz i wsp.32 opublikowali kryteria rozpoznawania LQTS, które zostały zmodyfikowane w 1993 roku i zawierają ważne wskazówki dotyczące wstępnej oceny potencjalnych przypadków. System ten wykorzystuje punktację od 1 do 9 opartą na wywiadzie rodzinnym oraz wynikach badań klinicznych i elektrokardiograficznych. Prawdopodobieństwo choroby jest małe przy wyniku ≥1, pośrednie przy 2-3, a duże przy ≥4 (tab. 2).

Diagnostyka prenatalna zespołu długiego QT
Bradykardia płodowa może być jedną z pierwszych manifestacji klinicznych LQTS. Retrospektywne serie badań wykazały, że do 70% pacjentów, u których w dzieciństwie rozpoznano LQTS, ma w wywiadzie bradykardię, zwykle z towarzyszącym wodogłowiem płodu.33 Ocena repolaryzacji serca płodu między 14. a 39. tygodniem jest przydatna we wczesnym rozpoznaniu LQTS.34
Mozaikowość gonad w kierunku LQTS wiąże się z powtarzającymi się utratami płodu w trzecim trymestrze ciąży.35 W przypadku dużego podejrzenia choroby amniopunkcja po 16 tygodniach ciąży może być przydatna w ustaleniu rozpoznania, które jest łatwo osiągalne, gdy wiadomo, że jedno z rodziców jest nosicielem określonej mutacji.36
BADANIE PACJENTA Z SYNDROMEM DŁUGIEGO QT
Historia kliniczna
Historia rodzinna i/lub osobista nagłego zgonu ma kluczowe znaczenie zarówno dla rozpoznania, jak i stratyfikacji ryzyka LQTS. Ponadto, czynniki poprzedzające i kontekst omdleń mogą wskazywać na podtyp LQTS. We wstępnej ocenie podejrzanego przypadku należy wykluczyć stosowanie leków, które mogą wydłużać odstęp QT.
QT Interval: What Is Normal?
Odstęp QT powinien być mierzony preferencyjnie w odprowadzeniach II lub V5,37 gdzie udowodniono, że ma większą wartość predykcyjną.38 Odstęp ten wskazuje czas trwania repolaryzacji komór i jest mierzony od początku załamka Q do końca załamka T. Konwencjonalnie stosuje się wzór zaproponowany przez Bazetta39 w celu skorygowania czasu trwania odstępu w zależności od częstości akcji serca (QTc=QT/√RR, wyrażony w sekundach). Chociaż pomiar odstępu QT wydaje się prosty, w wieloośrodkowym badaniu przeprowadzonym przez Viskina i wsp.40 mniej niż 40% lekarzy innych niż kardiolodzy, mniej niż 50% kardiologów i ponad 80% specjalistów zajmujących się arytmią wiedziało, jak prawidłowo go zmierzyć. Wskazane jest, aby lekarze wykonywali pomiar manualny i nie ufali pomiarom automatycznym, które mogą być przydatne w przypadku innych odstępów, ale są nieprecyzyjne przy obliczaniu odstępu QT. QT jest odstępem dynamicznym, a jego prawidłowa granica zależy od kilku czynników. Chociaż odstęp QTc wynoszący 440 ms u mężczyzn i 460 ms u kobiet jest uważany za nieprawidłowy, to w tym zakresie można znaleźć zarówno nosicieli mutacji, jak i osoby zdrowe (ryc. 3). W rodzinach z LQTS1 Vincent i wsp.41 wykazali, że żaden z przypadków z dodatnim genotypem nie miał QTc470 ms. Monnig i wsp.38 wykazali ostatnio, że QTc>440 ms wystarcza do wykrycia pacjentów z mutacjami związanymi z LQTS, QTc>470 ms jest przydatny do identyfikacji pacjentów zagrożonych wystąpieniem objawów, a QTc>500 ms występuje u objawowych pacjentów poddawanych leczeniu.
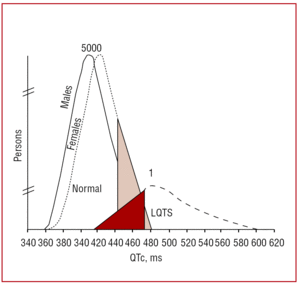
Rycina 3. Model przedstawiający rozkład odstępu QT skorygowanego częstością akcji serca (QTc) u pacjentów z mutacjami w KVLQT1, HERG lub SCN5A oraz u członków ich rodzin nie dotkniętych mutacjami. Krzywa po lewej stronie opisuje rozkład u niedotkniętych mutacją członków rodziny, a krzywa po prawej stronie u dotkniętych mutacją członków rodziny.
Inne zmiany elektrokardiograficzne związane z zespołem długiego QT
Pacjenci z LQTS mogą prezentować wiele zmian załamka T: między innymi naprzemienność biegunów, zmiany amplitudy, karbowanie i wygląd dwufazowy.42 Naprzemienność załamka T (ryc. 4A) definiuje się jako zmienność amplitudy, morfologii i polaryzacji załamka T w rytmie zatokowym, bez zmian w zespole QRS. Jest to wskaźnik niestabilności elektrycznej,43 odzwierciedlający regionalną dyspersję repolaryzacji komór, i czasami poprzedza migotanie komór.44
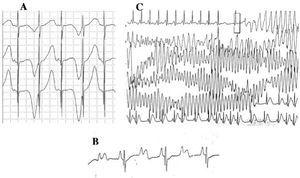
Ryc. 4. Zmiany elektrokardiograficzne w zespole długiego QT. A: alternans elektryczny załamka T. B: blok przedsionkowo-komorowy 2:1. C: samoograniczający się torsade de pointes.
U pacjentów z LQTS mogą wystąpić objawy dysfunkcji węzła zatokowego, bradykardia i/lub pauzy.45 Podtypy LQTS1 i LQTS3, zwłaszcza ten ostatni, często prezentują bradykardię zatokową,46 natomiast LQTS4 wiąże się z dysfunkcją węzła zatokowego.18
Od dekady lat 1970-1980 obserwuje się współistnienie wad przewodzenia AV z LQTS47 (ryc. 4B). Blok AV dwa do jednego jest rzadką manifestacją o złym rokowaniu, która może być obecna od okresu płodowego w postaci uporczywej bradykardii. Częstość występowania tej nieprawidłowości szacuje się na 4%-5%48 i wiąże się ona z dużą śmiertelnością pomimo leczenia beta-blokerami i/lub stymulatorami serca.49,50 Zjawisko to można wytłumaczyć długim czasem trwania potencjału czynnościowego. Gdy okres refrakcji komór ulega wydłużeniu, następujący po nim impuls czynności zatokowej jest blokowany, ponieważ dociera do komór, gdy znajdują się one jeszcze w okresie refrakcji. Ta zmiana wydaje się występować wyłącznie w LQTS, ponieważ okres refrakcji komór jest dłuższy niż okres refrakcji układu przewodzącego AV.51 Nachylenie zespołu QRS jest zwykle strome, a blok zlokalizowano w obszarze infraHis,46,51,52 ale miejsce bloku może zależeć od genotypu. Do tej pory 4 geny były związane z blokiem 2:1 w LQTS: HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 i SCN4B (LQTS10).55
Charakterystyczną arytmią komorową w LQTS jest torsade de pointes (ryc. 4C). Występuje w przypadku wydłużenia odstępu QT, niezależnie od etiologii. Jest to polimorficzny częstoskurcz komorowy spowodowany reentry, charakteryzujący się elektrokardiograficznie ciągłym skręcaniem osi QRS wokół urojonej linii. Często jest on poprzedzony pauzą, po której następuje ekstrasystolia (krótki-długi-krótki odstęp RR), jak pokazano na rycinie.56-58 Może zakończyć się migotaniem komór i nagłym zgonem. Jeśli do tego nie dojdzie, pacjent może doświadczyć jedynie omdlenia, a jeśli epizod jest krótki, może pozostać niewykryty.
Badanie holterowskie
Badanie holterowskie zapewnia pełną, dynamiczną ocenę odstępu QT. Sporadycznie rejestruje się spontaniczne epizody bezobjawowej arytmii komorowej, a także epizody dysfunkcji węzła zatokowego lub bloku AV.
Test wysiłkowy
Pacjenci z LQTS nie mogą osiągnąć maksymalnej oczekiwanej częstości akcji serca obliczonej w zależności od wieku. Ponadto przy wysiłku fizycznym odstęp QT może wykazywać paradoksalne zachowanie, zwiększając się zamiast zmniejszać.59,60 Obraz elektrokardiograficzny podczas wysiłkowej próby wysiłkowej będzie różny w zależności od typu LQTS. Pacjenci z LQTS1, oprócz tego, że nie osiągają maksymalnej obliczonej dla ich wieku częstości rytmu serca, często wykazują wydłużenie odstępu QT, podczas gdy pacjenci z LQTS2 mogą osiągać oczekiwaną częstość rytmu serca i wykazywać jedynie łagodne wydłużenie odstępu QT lub nie wykazywać go wcale.61,62 Ogólnie rzecz biorąc, pacjenci z LQTS3 wykazują fizjologiczną odpowiedź na wysiłek fizyczny, tj. prawidłowe skrócenie odstępu QT.63 Testy wysiłkowe mogą być również przydatne w ocenie odpowiedzi na leczenie i stratyfikacji ryzyka w przypadkach bezobjawowych lub gdy istnieją wątpliwości co do zdarzeń prowadzących do arytmii.
Przesiewowe badania genetyczne
W ostatnich latach badania genetyczne w LQTS ograniczały się do laboratoriów badawczych. Niemniej jednak informacje uzyskane dzięki tym badaniom okazały się niezwykle przydatne w leczeniu pacjentów, szczególnie tych z grupy wysokiego ryzyka. Być może głównym zastosowaniem badań przesiewowych jest poradnictwo genetyczne, ale mają one również istotne implikacje w leczeniu, które może być ukierunkowane w zależności od dotkniętego kanału. Dokładna lokalizacja danej mutacji może dostarczyć dodatkowych informacji na temat ewolucji ryzyka. Pacjenci z mutacjami w regionie transmembranowym KCNQ1 (IKS) mają większe prawdopodobieństwo wystąpienia zdarzeń arytmicznych niż ci z mutacjami w regionie C-końcowym64; to samo dotyczy pacjentów z mutacjami w regionie porowym KCNH2 lub HERG65 w porównaniu z tymi z mutacjami w regionie N- lub C-końcowym66.
Wstępne badania przesiewowe można być może ograniczyć do genów KCNQ1, HERG i SCN5A, które dają możliwość napotkania mutacji w 65% przypadków. Gdy uzyskane wyniki są negatywne, badania przesiewowe można rozszerzyć o geny KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 i SCN4B, co zwiększy możliwość uzyskania pozytywnych wyników o 5% do 10%.
Pośmiertne badania genetyczne
Ciekawe jest to, że mutacje genów prowadzące do LQTS znaleziono u dzieci, które doświadczyły nagłej śmierci oraz w niewytłumaczalnych przypadkach nagłej śmierci u młodych dorosłych.
Pośmiertne badania genetyczne pacjentów z nagłym zgonem i negatywnym wynikiem autopsji wykazały mutacje prowadzące do LQTS w różnym odsetku67-69: blisko 10% u dzieci i 35% u młodych dorosłych.70-72 W oparciu o te wyniki zaproponowano rutynowe badanie EKG u wszystkich noworodków.73,74
Pośmiertne badania genetyczne, znane w literaturze jako „molekularna autopsja”, oprócz reperkusji prawnych, mają ważne implikacje dla rodzin, w których mogą występować zmiany, a które o tym nie wiedzą.
Polimorfizmy regulacyjne
W populacji LQTS opisano kilka często występujących polimorfizmów, rozmieszczonych w prawie wszystkich genach związanych z tym schorzeniem. Chociaż zmiany te najwyraźniej nie są patogenne, niektóre z nich mogą mieć następujące skutki75-78:
1. Generować indywidualną podatność na rozwój arytmii.
2. Sprzyjać patogennemu wpływowi innej zmiany nonsynonimicznej.
3. Zmniejszać patogenny wpływ innej zmiany nonsynonimicznej.
Tak jest w przypadku polimorfizmu K897T w KCNH2 (HERG), który występuje nawet u 15% populacji i jest nie tylko związany z podatnością na niektóre leki,79 ale także sprzyja patogennemu wpływowi mutacji w tym samym genie.78. Innym przykładem jest polimorfizm S1103Y w genie SCN5A, występujący głównie u osób rasy czarnej, którego częstość występowania wynosi blisko 13% i wiąże się ze zwiększonym ryzykiem nagłego zgonu w dzieciństwie.80
Co ciekawe, w produkcie genu SCN5A, (który koduje izoformę kanału sodowego Nav1,5 u ludzi) opisano dwa alternatywne miejsca przetwarzania generujące dwa typy kanałów sodowych: jedno z 2016 aminokwasami zawierającymi glutaminę w pozycji 1077 (Q1077), a drugie z 2015 aminokwasami pozbawionymi glutaminy (Q1077del). Transkrypty tych alternatywnych procesów są obecne w proporcji 2:1 w tym samym ludzkim sercu i kilka częstych polimorfizmów będzie miało różny wpływ na funkcjonowanie kanału, w zależności od tego, czy kontekstem jest Q1077 czy Q1077del. Zostało to początkowo wykazane w przypadku polimorfizmu H558R SCN5A, występującego nawet u 30% populacji. Gdy H558R ulegał ekspresji w kontekście Q1077, obserwowano głęboką redukcję prądu jonowego.81 Podobny efekt udokumentowano w przypadku polimorfizmu S524Y82. Wyniki te dostarczyły czynników wyjaśniających zróżnicowane nasilenie choroby, a także różne fenotypy tej samej mutacji obserwowane w niektórych rodzinach.77
Testy farmakologiczne z adrenaliną
Testy farmakologiczne z małą dawką adrenaliny są bezpieczną, przydatną opcją demaskowania podejrzanych przypadków LQTS z granicznym odstępem QTc. Jest ona szczególnie skuteczna w wykrywaniu bezobjawowych postaci LQTS1, z czułością 92,5%, swoistością 86%, dodatnią wartością predykcyjną 76% i ujemną wartością predykcyjną 96%. Może być również przydatna w diagnostyce LQTS2, ale z mniejszą czułością i swoistością. Nie jest przydatna w przypadku LQTS3 i innych postaci LQTS. W warunkach prawidłowych stymulacja współczulna indukuje fosforylację kanału potasowego IKs, optymalizując jego funkcję i powodując skrócenie potencjału czynnościowego. U pacjentów z LQTS, zwłaszcza typu 1, obserwuje się paradoksalną reakcję na podanie adrenaliny w małej dawce (0,025-0,2 µg/kg/min), która wydłuża odstęp QT do ponad 30 ms83-86.
QT INTERVAL PROLONGATION AND DRUG-INDUCED TORSADE DE POINTES
Duża różnorodność leków stosowanych w różnych specjalnościach medycznych może powodować jatrogenne wydłużenie odstępu QT. Niektóre leki zostały wycofane z rynku z powodu tego niepożądanego działania (m.in. astemizol i cyzapryd; więcej informacji można znaleźć na stronie www.qtdrugs.org).87,88
Komorowe zaburzenia rytmu serca wtórne do stosowania leków nieantyarytmicznych występują u mniej niż jednego z każdych 10 000 do 100 000 narażonych osób. Biorąc pod uwagę, że badania kliniczne obejmują od 2000 do 3000 osób, to niepożądane i śmiertelne zdarzenie niepożądane mogłoby łatwo umknąć wykryciu podczas fazy klinicznej opracowywania leku.89 Ten punkt wywołał ogromne zainteresowanie aspektami związanymi z bezpieczeństwem badań i rozwoju nowych leków.
Do czynników związanych z indywidualną podatnością należą: płeć żeńska, hipokalcemia, hipomagnezemia, bradykardia, niewydolność serca, stan po kardiowersji, migotanie przedsionków, przerost lewej komory, niewykryty LQTS, predysponujące polimorfizmy i duże stężenia predysponujących leków w surowicy.90
Kanałem, który zazwyczaj wchodzi w interakcje z lekami, jest IKr, kodowany przez gen KCNH2(HERG), ze względu na swoją budowę molekularną. Inne kanały potasowe mają 2 reszty prolinowe skierowane pod kątem w stronę porów kanału, zmniejszając jego światło. W przeciwieństwie do IKr brak tych reszt powoduje powstanie większego przedsionka porów i ułatwia ekspozycję na duże cząsteczki. Ponadto posiada 2 reszty aromatyczne (tyrozynę i fenyloalaninę), które sprzyjają wiązaniu się z cząsteczkami aromatycznymi obecnymi w kilku lekach zdolnych do blokowania kanału.91
Jak wspomniano powyżej, penetracja LQTS jest niepełna i niektórzy bezobjawowi nosiciele mutacji mogą manifestować złośliwą arytmię po otrzymaniu jednego z tych leków. Ponadto, polimorfizmy uważane za częste w populacji nadają indywidualną podatność na rozwój torsade de pointes w przypadku stosowania niektórych leków. Tak jest w przypadku polimorfizmu R1047L, drugiego co do częstości występowania w genie KCNH2, który wiązano z rozwojem torsade de pointes przy stosowaniu leku dofetylid.92 U osób zdrowych opisano co najmniej 20 polimorfizmów genu KCNH2, a ich wpływ na indywidualną podatność na wystąpienie polekowej arytmii pozostaje do ustalenia.93 Polimorfizmy zwiększające podatność na rozwój arytmii komorowej udokumentowano również w kanale sodowym Na1.5. Dotyczy to polimorfizmu H558R, który występuje nawet u 30% populacji, czy S1103Y, który jest częsty u osób rasy czarnej80,81,90,94,95; Nie zbadano ich wpływu na wrażliwość na leki.
DŁUGI SYNDROM QT A CIĄŻA
Poradnictwo genetyczne jest ważne w LQTS, ale ogólnie rzecz biorąc nie ma przeciwwskazań do zajścia w ciążę u kobiet będących nosicielkami, chociaż każdy przypadek jest inny i powinien być oceniany indywidualnie w odpowiednim kontekście.
Zauważono, że ryzyko wystąpienia złośliwej arytmii komorowej zmniejsza się wraz z ciążą. Z kolei większą podatność na wystąpienie złośliwej arytmii odnotowano w ciągu pierwszych 9 miesięcy po porodzie, zwłaszcza u pacjentek z LQTS2. Ryzyko to zmniejsza się znacznie po zastosowaniu terapii beta-blokerami.96
ZNACZENIE RYZYKA
Ewolucja LQTS jest zróżnicowana i zależy od czasu trwania odstępu QTc, czynników środowiskowych, wieku, genotypu i odpowiedzi na leczenie.97,98 Komorowe zaburzenia rytmu serca występują częściej w LQTS1 i LQTS2, ale są bardziej nasilone w LQTS3.99 Jak wspomniano powyżej, kobiety są szczególnie podatne na wystąpienie złośliwej arytmii w okresie poporodowym.14
Zespół długiego QT należy uznać za zespół wysokiego ryzyka, gdy towarzyszy mu:
1. Wrodzona głuchota (zespół Jervella-Lange-Nielsena).
2. Nawracające omdlenia spowodowane złośliwą tachyarytmią komorową.
3. Nagły zgon w wywiadzie rodzinnym.
4. QTc>500 ms.
5. Blok przedsionkowo-komorowy 2:1.
6. Alternans elektryczny załamka T.
7. Genotyp LQTS3.
W badaniu Priori i wsp.97 przeprowadzonym u 647 pacjentów wykazano, że prawdopodobieństwo wystąpienia poważnego zdarzenia (omdlenia, zatrzymanie krążenia, nagły zgon) przed 40. rokiem życia jest wysokie (>50%), gdy QTc wynosi >500 ms w LQTS1, LQTS2 oraz u mężczyzn z LQTS3. Ostatnio przeprowadzono analizę międzynarodowego rejestru LQTS. Analizowano ryzyko nagłego zgonu u 2772 nastolatków z tą chorobą i zidentyfikowano 3 czynniki związane z większym ryzykiem w tej populacji: QTc>530 ms, omdlenia w wywiadzie w ciągu ostatnich 10 lat oraz płeć; 10-12-letni chłopcy mieli większe ryzyko niż dziewczęta, ale w przedziale wiekowym 13-20 lat ryzyko było porównywalne.100
LECZENIE
U pacjentów objawowych, którzy nie otrzymują leczenia, śmiertelność roczna wynosi 20%, a śmiertelność 10-letnia 50% po pierwszym zdarzeniu arytmii komorowej. Chociaż jest oczywiste, że leczenie należy rozpoczynać w przypadku wystąpienia objawów, nadal dyskutuje się na temat podejścia, jakie należy stosować u pacjentów bezobjawowych. Udokumentowano, że zatrzymanie krążenia może być pierwszą manifestacją choroby u 9% pacjentów,48 a u 12% pacjentów bezobjawowych wystąpią objawy i może dojść do nagłego zgonu. U wszystkich pacjentów z LQTS należy rozpocząć leczenie beta-blokerami. Zalecane jest ograniczenie wysiłku fizycznego, ale kliniczne i elektrokardiograficzne markery ryzyka są przydatną podstawą do podjęcia decyzji. Ważne jest, aby poinformować pacjentów o ryzyku stosowania kilku leków, które mogą wydłużać odstęp QT i sprzyjać rozwojowi arytmii komorowej, o czym wspomniano powyżej. Diagnostyka genetyczna, oprócz umożliwienia odpowiedniego poradnictwa rodzinnego związanego z chorobą, jest pomocna w ocenie rokowania i ukierunkowaniu specyficznego leczenia.
Beta-blokery
Beta-blokery są lekami pierwszego rzutu w LQTS i wszyscy pacjenci powinni je otrzymywać jako terapię początkową.101 Zapewniają one zmniejszenie ryzyka zdarzeń sercowo-naczyniowych nawet o 64%100 i są szczególnie skuteczne u pacjentów z mutacjami kanałów IKs (LQTS1),102 które są w dużym stopniu regulowane przez układ współczulny. Beta-blokery nie modyfikują odstępu QT, lecz jego dyspersję.103 Mimo że leki te zmniejszają częstość występowania zdarzeń,104,105 wykazano, że 10% pacjentów z LQTS1, 23% z LQTS2 i 32% z LQTS3 będzie miało objawy sercowo-naczyniowe mimo leczenia.106 Szczególnie u pacjentów z LQTS3 nie wydaje się możliwe uzyskanie istotnych korzyści; w rzeczywistości u tych chorych należy ostrożnie stosować tę grupę leków, ponieważ epizody komorowych zaburzeń rytmu serca w LQTS3 występują częściej, gdy częstość akcji serca jest mała. Ogólnie rzecz biorąc, u 32% pacjentów z objawami wystąpi nawrót objawów w ciągu pierwszych 5 lat przed rozpoczęciem leczenia beta-blokerami, a u 14% pacjentów uratowanych po epizodzie nagłej śmierci wystąpi kolejne podobne zdarzenie w ciągu 5 lat, jeśli będą oni otrzymywać tylko tę terapię.107 W leczeniu LQTS stosowano kilka beta-blokerów, głównie nadolol (0,5-1 mg/kg/dobę), propranolol (2-4 mg/kg/dobę), metoprolol (0,5-1 mg/kg/dobę) i atenolol (0,5-1 mg/kg/dobę). Atenolol może jednak nie być korzystny w LQTS; poinformowano, że co najmniej 75% pacjentów, u których nie uzyskano odpowiedzi na leczenie beta-blokerami, otrzymywało atenolol, chociaż stwierdzenie to może być związane ze stosowaniem suboptymalnych dawek.104 Próba wysiłkowa jest przydatna do ustalenia odpowiedniej dawki. Maksymalna częstość akcji serca nie powinna przekraczać 130 uderzeń/min podczas leczenia.
Blokery kanału sodowego
Mutacje kanału sodowego, które powodują LQTS3, powodują wadliwą inaktywację kanału; blokada kanału sodowego okazała się przydatna u tych pacjentów. W badaniach z zastosowaniem flekainidu udokumentowano poprawę częstości akcji serca, zmian załamka T i odstępu QT.108 Zgłaszano również, że meksyletyna poprawia elektrokardiograficzne markery ryzyka.63,109,110 W badaniach in vitro z zastosowaniem ranolazyny wykazano zmniejszenie szkodliwych skutków mutacji opisywanych u ludzi.111 Chociaż wyniki te są zachęcające, należy pamiętać, że nie ma długoterminowych badań oceniających tę terapię ani wyników dużych serii. Blokery kanałów sodowych nie powinny być podawane w przypadku braku potwierdzonego rozpoznania genetycznego.
Suplementacja potasu i leki zwiększające jego dostępność
Suplementacja potasu i/lub leki oszczędzające potas, takie jak spironolakton, skracają odstęp QTc w 24% przypadków.112,113 Leki sprzyjające otwieraniu kanałów potasowych, takie jak aprikalim, lewokromakalim, nikorandil i pinacidil, okazały się przydatne w leczeniu LQTS. Podtypami, w których odnoszą one szczególną korzyść, są LQTS1 i LQTS2.114
Stymulatory serca i defibrylatory
Stymulacja stymulatora serca była stosowana u pacjentów z arytmią zależną od pauzy.115,116 Pacjenci z LQTS3 zwykle odnoszą większe korzyści z tego leczenia, ponieważ w tej grupie częstość występowania bradykardii jest większa. Stymulacja DDD jest wskazana u pacjentów z arytmią zależną od pauzy lub blokiem AV wysokiego stopnia 2:1. Częstotliwości zaprogramowane poniżej 70 uderzeń/min117 nie są przydatne w zapobieganiu arytmii komorowej. Zaleca się zaprogramowanie czujnika na szybką odpowiedź, ponieważ u tych pacjentów zwykle występuje nieodpowiednie przyspieszenie rytmu serca w odpowiedzi na wysiłek fizyczny. Należy wyłączyć wszystkie funkcje, które sugerują obecność pauz, takie jak histereza i funkcja nocna. PARP (postventricular atrial refractory period) powinien być tak krótki, jak to tylko możliwe. Funkcja regulacji częstotliwości powinna być włączona, aby zapobiec pauzie ekstrasystolicznej. Należy pamiętać, że oversensing załamka T i błędy wychwytu również mogą być przyczyną pauz. Łączne stosowanie wszczepialnego kardiowertera-defibrylatora (ICD) i beta-blokerów istotnie zmniejsza częstość występowania nagłego zgonu.118-120 Wskazania do stosowania tych środków są oczywiste w przypadkach wysokiego ryzyka.121 Programowanie urządzenia będzie się różnić w zależności od potrzeb konkretnego pacjenta, ale generalnie należy unikać podawania leczenia w bezobjawowych, samoograniczających się zdarzeniach; w tym celu wskazany jest czas detekcji wynoszący 15 s. Powikłaniem terapii AID jest burza arytmiczna. U blisko 15% pacjentów może wystąpić to powikłanie, które wynika w dużej mierze ze zwiększonego napięcia współczulnego po wstrząsie ICD.118 Problem ten można opanować poprzez zwiększenie dawki beta-blokera. Jeśli ten sposób postępowania okaże się nieskuteczny, należy rozważyć resekcję zwojów łańcucha współczulnego.
Lewa sympatektomia
W 1971 roku wprowadzono zwoje współczulne jako przydatną opcję terapeutyczną u tych pacjentów.122 W 1991 roku Schwartz i wsp.123 opublikowali pierwszą serię 85 pacjentów słabo reagujących na leczenie beta-blokerami, u których wykonano lewą stellektomię z zachęcającymi wynikami: 5-letnie przeżycie wyniosło 94%. Obecnie tę opcję terapeutyczną proponuje się chorym z grupy wysokiego ryzyka, u których omdlenia utrzymują się mimo leczenia beta-blokerami i/lub wszczepienia stymulatora serca, a także tym, którzy doświadczają częstych wstrząsów z wszczepionego defibrylatora. Zabieg polega na resekcji dolnej części zwoju gwiaździstego i zwojów T2 do T4 lewego zwoju piersiowego łańcucha współczulnego, ponieważ zwykła stellectomia lewostronna nie okazała się wystarczająco skuteczna. Z dobrym skutkiem stosowano torakoskopię mikroinwazyjną124,125 . Niedawno opublikowano największą serię chorych leczonych tą metodą, w której wykazano istotne zmniejszenie liczby epizodów omdleń lub nagłych zgonów, a także 5-letnie przeżycie wynoszące 95%. U pacjentów z wcześniejszymi omdleniami 5-letnie przeżycie wynosiło 97%, z 11% prawdopodobieństwem nawrotu, który w większości przypadków polegał na pojedynczym zdarzeniu synkopalnym. Stwierdzono również istotne skrócenie odcinka QT po lewostronnej sympatektomii. Mimo tak korzystnych wyników, zapobieganie nagłym zgonom nie jest całkowite, ale zostało zredukowane do 3%. U chorych z ICD, u których wykonano zabieg z powodu wielokrotnych wstrząsów defibrylatora, średnia liczba zdarzeń zmniejszyła się z 25 do 0, co stanowi redukcję o 95%. Korzystny efekt potwierdzono w LQTS1. Korzyści są prawdopodobnie mniejsze u pacjentów z LQTS2, a w LQTS3 nie udowodniono jego skuteczności.126
Ablacja
Donoszono, że ablacja ekstrasystolii, która w niektórych przypadkach inicjuje arytmię komorową, może być przeprowadzona ze zmniejszeniem częstości występowania epizodów.127 Nie ma jednak długoterminowych badań z odpowiednią liczbą pacjentów, które uzasadniałyby rutynowe stosowanie tej techniki.
Patrz artykuł redakcyjny na stronach 675-82
SKRÓTY
AV: atrioventricular
AID: automatic implantable defibrillator
ECG: elektrokardiogram
QTc: heart rate-corrected QT
ATS: zespół Andersena-Tawila
LQTS: zespół długiego QT
Dr Medeiros otrzymuje wsparcie ekonomiczne od CONACyT i FUNSALUD.
.