INTRODUÇÃO
Síndrome do QT Longo (SQTL) é caracterizada por uma repolarização ventricular gravemente alterada, resultando no prolongamento do intervalo QT no eletrocardiograma (ECG). A condição predispõe os pacientes a arritmia ventricular maligna (torsade de pointes) e morte súbita. A descrição clínica e eletrocardiográfica da síndrome do QT longo foi relatada em 1957 por Anton Jervell e Fred Lange Nielsen1, que publicaram seus estudos sobre uma família de pais não consanguíneos com 6 filhos. Quatro das crianças tiveram surdez congênita e episódios sincopais, e 3 apresentaram morte súbita. O estudo de ECG desses pacientes mostrou um intervalo QT anormalmente longo. Ambos os pais eram assintomáticos, tinham um ECG normal, e não apresentavam problemas auditivos. Em 1964, Romano e Ward relataram independentemente uma síndrome cardíaca caracterizada por síncope recorrente, história familiar de morte súbita e prolongamento do intervalo QT sem surdez neuronal.2 Estudos genéticos posteriores mostraram que a síndrome descrita por Jervell e Lange Nielsen, que é acompanhada por surdez neuronal congênita, corresponde a mutações homozigotos, com fenótipo grave e alto risco de morte súbita. A condição conhecida como síndrome Romano-Ward corresponde geralmente a mutações heterozigotas, os pacientes não apresentam alterações auditivas e a gravidade da doença varia consideravelmente. Quase meio século depois, em 1995,3,4 os principais genes associados à SQTL foram descritos e a doença foi reconhecida como um distúrbio do canal iônico cardíaco. Foi a primeira canaleopatia cardíaca a ser descrita e é talvez o distúrbio do canal iônico arritmogênico mais investigado até hoje. O quadro clínico varia muito: o paciente pode ser assintomático, ou mostrar síncope recorrente, convulsões, ou morte súbita como primeira manifestação da doença. Inicialmente, a SQTL foi considerada uma síndrome rara e, na verdade, a apresentação grave da doença é esporádica. No entanto, a incidência de mutações relacionadas é estimada em 1/3000-5000 casos,5 32% dos portadores assintomáticos podem ter um intervalo QT corrigido pela freqüência cardíaca (QTc) dentro dos limites normais, a doença é transmitida a 50% de seus descendentes, eles são mais suscetíveis a desenvolver arritmia quando comparados à população geral, e até 20% podem tornar-se sintomáticos.6
A síndrome QTc longa exibe grande heterogeneidade genética. Mais de 500 mutações, distribuídas em 10 genes, foram descritas nesta condição: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3, e SCN4B. Apesar dos avanços nesta área, o diagnóstico genético não pode ser estabelecido em 25%-30% dos pacientes.7,8 A apresentação da doença é principalmente monogênica6; as variedades poligênicas ou compostas geralmente têm um fenótipo mais severo. Penetrance, ou seja, pacientes que têm a mutação e manifestam o fenótipo, varia de 25% a 90%.9 Com menor freqüência, pode haver variações na expressividade da doença, com vários fenótipos resultantes da mesma mutação. Os estudos genéticos moleculares desenvolvidos nos últimos 11 anos produziram importantes correlações genótipo-fenótipo, que ajudaram a orientar a abordagem do tratamento. Além disso, observações interessantes têm sido feitas sobre a suscetibilidade individual ao desenvolvimento de arritmias em estudos que investigam os freqüentes polimorfismos não-sinônimos nesta população, um aspecto que tem despertado considerável interesse, particularmente na área da farmacogenômica.
CLASSIFICAÇÃO DA SÍNDROME QT LONGA
Conceitos Gerais
A classificação da SQTL utilizada no passado foi baseada na apresentação homozigota ou heterozigota da doença, que dá origem à síndrome de Jervell-Lange-Nielsen (com surdez) e à síndrome de Romano-Ward (sem surdez), respectivamente. A presente classificação enfatiza os achados genéticos, como é ilustrado na Tabela 1. Os 3 principais genes associados com a doença foram descritos em 1995-1996. Estes genes, que codificam as unidades porosas dos canais de potássio IKs e IKr, e o canal de sódio Nav1.5, são responsáveis por quase 65% dos casos. Embora nos anos seguintes sete genes adicionais tenham sido incluídos na lista, eles representam apenas 5% dos casos.
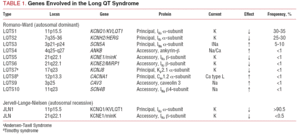
Os canais de íons são proteínas transmembranas que transportam íons através da membrana celular. Os canais implicados na SQTL são seletivos ou especializados no transporte de um único íon e são dependentes da tensão, ou seja, sua ativação ocorre em uma tensão intracelular específica, que varia de acordo com o subtipo do canal. Os fenômenos elétricos e contráteis que ocorrem no cardiomiócito são controlados por essas estruturas. Os canais de íons formam complexos macromoleculares que consistem em uma unidade principal que forma o poro do canal e proteínas auxiliares que o regulam (Figura 1). A disfunção do canal observada na SQTL pode ocorrer nestes dois locais: a proteína principal ou as proteínas reguladoras (Tabela 1). O envolvimento da unidade formadora de poros, conhecida como alfa, gera os três subtipos mais comuns da SQTL: LQTS1 (afetando o canal de potássio IKs), LQTS2 (afetando o canal de potássio IKr), e LQTS3 (afetando o canal de sódio). Como estes são os subtipos mais frequentes, são os mais caracterizados clínica e geneticamente. As correlações fenótipo-genótipo nestas três formas principais estão descritas na Figura 2. Atualmente, a síndrome de Jervell-Lange-Nielsen corresponde às variedades da SQTL 1 e 5. Caracteristicamente, estes pacientes apresentam surdez congénita e mutações homozigotas ou heterozigotas compostas que afectam a corrente IKs. A síndrome de Romano-Ward inclui variedades da SQTL 1 a 10 e não envolve surdez.
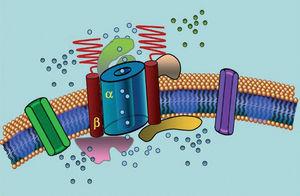
Figure 1. Representação esquemática do complexo macromolecular. Os canais iônicos são proteínas transmembranas (α) reguladas por várias proteínas; uma delas é a chamada subunidade β.
Síndrome do QT Longo Tipo 1 (SQTL1)
Pacientes com SQTL1 geralmente apresentam episódios de arritmia ventricular quando se exercitam ou quando passam por estímulo simpático (68%).10 Nadar tem sido descrito como um esporte desencadeando arritmia na SQTL1.11 Penetrance é quase 62% neste subtipo. A onda T nesses pacientes frequentemente tem uma base ampla e duração muito prolongada12,13 (Figura 2). É o subtipo mais freqüente e explica 30%-35% dos casos. O gene afetado, KvLQT1 (ou KCNQ1), está localizado no cromossomo 11 (11p15,5) e códigos para o canal de potássio IKs α-subunit. O potencial de ação é prolongado por uma redução na corrente K+ de saída durante a fase 3 do potencial de ação.
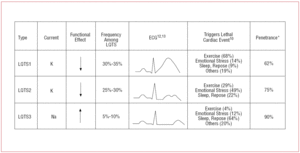
Figure 2. Correlação genótipo-fenótipo nas síndromes QT longas mais frequentes. *Refere a casos que têm a mutação e manifestam o fenótipo.
Síndrome do QT Longo Tipo 2 (SQTL2)
Pacientes com SQTL2 tendem a apresentar arritmia ventricular em resposta a estresse emocional (49%) ou estímulos auditivos súbitos (por exemplo, um alarme-relógio), e menos freqüentemente durante o sono (22%) ou exercício (29%).10 Mulheres no período pós-parto são particularmente suscetíveis.14 A penetração estimada é de 79%; portanto, até 20% dos casos podem ter um ECG não-diagnóstico. A onda T na SQTL2 é geralmente de baixa amplitude e bífida, com entalhes12,13 (Figura 2). O gene afetado é KCNH2 ou HERG, localizado no cromossomo 7 (7q35-36), que codifica o canal de potássio IKr α-subunit e responde por 25%-30% dos casos. A disfunção deste canal diminui a corrente K+ de saída durante a fase 3 do potencial de ação, prolongando sua duração.
Síndrome do QT Longo Tipo 3 (LQTS3)
Pacientes com LQTS3 têm um risco maior de apresentar arritmias malignas durante o repouso (sono) ou bradicardia.15 A penetração da mutação do gene SCN5A é de quase 90%. O ECG na SQTL3 geralmente mostra uma onda T atrasada e pontiaguda e permite uma clara observação do prolongamento do segmento ST12,13 (Figura 2). Esses pacientes geralmente apresentam menos sintomas que aqueles com SQTL1 ou SQTL2, mas os eventos são caracteristicamente mais letais.
O gene afetado na SQTL3 é o SCN5A, que codifica o canal de sódio Nav1,5 α-subunit (Figura 1), localizado no cromossomo 3 (3p21-24); é a causa da doença em 5%-10% dos casos. A inativação defeituosa do canal permite a entrada sustentada de Na+ durante a fase 2 do potencial de ação, prolongando sua duração.
Síndrome do QT Longo Tipo 4 (SQTL4)
O Tipo 4 é uma variedade rara de SQTL, sendo responsável por quase 1% dos casos. É uma forma atípica que produz um amplo espectro de arritmias, incluindo taquicardia ventricular polimórfica catecolaminérgica, fibrilação atrial, alterações da condução intraventricular, disfunção dos nós sinusais e bradicardia6-18; além disso, o QTc pode estar dentro dos limites normais em muitos pacientes. O gene afetado é o ANKB, localizado no cromossomo 4 (4q25-27), que codifica a síntese da anquilina-β, uma proteína estrutural que liga as proteínas da membrana cardiomiocitária às proteínas do citoesqueleto. Estas proteínas são a bomba de Na/K ATPase, o permutador Na/Ca e o receptor de trifosfato de inositol (InsP3R). As mutações que causam uma perda da função anquilina-β levam a aumentos na concentração de cálcio intracelular e alterações na expressão da N/K ATPase e permutador de Na/Ca. A elevada concentração de cálcio dá origem a uma pós-despolarização precoce e retardada. Assim, as arritmias ventriculares observadas nas mutações do gene anquilina-β são devidas a despolarizações espontâneas, geralmente em resposta à estimulação catecolaminérgica.
Síndrome do QT Longo Tipo 5 (LQTS5)
Tipo 5 origina-se com alterações na seqüência do gene KCNE1 localizado no cromossomo 21 (21q22.1p22.)19 códigos KCNE1 para síntese do canal IKs β-subunit, também conhecido como subunidade minK, que regula o canal IKs. Este tipo representa menos de 1% dos casos.
Síndrome do QT Longo Tipo 6 (LQTS6)
O gene afetado no tipo 6 é KCNE2, localizado no cromossomo 21 (21q22.1).20 Este gene codifica o canal de potássio β-subunit, também conhecido como subunidade MiRP1, e regula o canal IKr. Menos de 1% dos casos são do tipo 6.
Síndrome do QT Longo Tipo 7 ou Síndrome de Andersen-Tawil (LQTS7)
Os achados dismórficos e alterações eletrocardiográficas observadas nesta síndrome foram descritos pela primeira vez em 1971 pelo Dr. Andersen21 e revisados em 1994 pelo Dr. Tawil,22 mas a descrição genética/molecular não foi relatada até 2001.Atualmente conhecida como síndrome de Andersen-Tawil (ATS), essa condição é uma alteração autossômica dominante caracterizada por paralisia periódica, desenvolvimento esquelético anormal, arritmia ventricular do tipo envolvendo extra-sístoles ventriculares freqüentes e uma suscetibilidade particular ao desenvolvimento de fibrilação ventricular, particularmente em mulheres. As alterações descritas na ATS incluem extra-sístoles ventriculares (41%), taquicardia ventricular polimórfica não sustentada (23%), taquicardia ventricular bidirecional (68%) e torsade de pointes (3%).24 Algumas das características dismórficas observadas incluem baixa estatura, escoliose, clinodactilia, hipertelorismo, baixo implante das orelhas, micrognatia e fronte larga. A expressão da doença varia, fato que complica o diagnóstico precoce.23,25 Mutações no gene KCNJ2 localizado no cromossomo 17 (17q23), que codifica a síntese do canal retificador de potássio Kir 2.1, responde por 70% dos casos. Este canal participa da fase 4 do potencial de ação. Vários autores questionam a inclusão deste gene no grupo causal da SQTL, pois o intervalo QTc é apenas ligeiramente prolongado nesta síndrome ou mesmo normal, mas a onda U é geralmente proeminente, o que levou à superestimação do intervalo QT. O leitor vai descobrir que alguns autores sugerem que as mutações do KCNJ2 geram ATS1 e não LQTS7,24
Síndrome do QT longo Tipo 8 (LQTS8)
Tipo 8 surge de mutações no gene CACNA1 localizado no cromossomo 12 (12p13,3), que codifica o Cav1 do canal de cálcio do tipo L.Causa síndrome de Timothy,26 uma condição caracterizada por malformações cardíacas, deficiência imunológica intermitente, hipoglicemia, alterações cognitivas incluindo autismo, fusão interdigital e QT prolongado, o que leva a arritmia cardíaca e morte súbita.27 Menos de 0,5% dos casos são do tipo 8,
Síndrome do QT Longo Tipo 9 (SQTL9)
Esta variedade de SQTL desenvolve-se a partir de mutações no gene CAV3, localizado no cromossomo 3 (3p25), que codifica a síntese da caveolina 3. A caveola é uma invaginação da membrana plasmática implicada na endocitose, homeostase lipídica e transdução de sinal. Um componente importante desta estrutura é a caveolina, que tem 3 subtipos conhecidos; o subtipo 3 é específico para o músculo esquelético e cardíaco. Alguns canais iônicos são co-localizados na caveola, incluindo uma isoforma cardíaca do canal de sódio Nav1,5. Várias mutações nesta proteína foram descritas recentemente. Estas alteram as propriedades biofísicas do canal de sódio Nav1,5 in vitro, gerando um fenótipo semelhante ao observado na SQTL3,28 Menos de 1% dos casos são atribuídos a esta causa.
Síndrome do QT Longo Tipo 10 (SQTL10)
Tipo 10 foi descrito em um caso muito grave, com QTc >600 ms, bradicardia fetal, e bloqueio atrioventricular (AV) 2:1. É resultante de mutações no gene SCN4B, localizado no cromossomo 11 (11q23), que codifica o canal de sódio β4-subunit. Foram descritos quatro subtipos diferentes de subunidades β, que interagem e regulam as várias isoformas do canal de sódio; no entanto, até agora, apenas o subtipo 4 foi associado à arritmogênese.29 A incidência de mutações deste gene não foi examinada, mas é estimada em
Mutações da variedade Jervell-Lange-Nielsen
Esta forma grave de SQTL é causada por mutações homozigotas30 ou heterozigotas compostas dos genes KCNQ1, e/ou KCNE1, que codificam a corrente IKs; ou seja, uma variedade muito grave das formas LQTS1 ou LQTS5. Esta condição é caracteristicamente associada com surdez congênita. Os pacientes geralmente têm um QTc>500 ms e síncope recorrente, e estão em alto risco de morte súbita. Os pais dos pacientes com essa variedade são geralmente heterozigotos e têm doença menos grave, ou não apresentam sintomas.31
DIAGNÓSTICO DE SÍNDROME QT LONGO
Swartz Score
Em 1985, Schwartz et al32 publicaram os critérios para diagnóstico da SQTL, que foram modificados em 1993 e contêm diretrizes importantes para a avaliação inicial de casos potenciais. Este sistema utiliza uma pontuação de 1 a 9 com base na história familiar e nos achados clínicos e eletrocardiográficos. A probabilidade de doença é baixa em uma pontuação de ≥1, intermediária em 2-3, e alta em ≥4 (Tabela 2).

Diagnóstico pré-natal da Síndrome do QT Longo
Bradicardia fetal pode ser uma das primeiras manifestações clínicas da SQTL. Séries retrospectivas têm mostrado que até 70% dos pacientes diagnosticados com SQTL durante a infância têm história de bradicardia, geralmente acompanhada de hidropisia fetal.33 A avaliação da repolarização cardíaca fetal entre as semanas 14 e 39 é útil para o diagnóstico precoce da SQTL.34
O mosaicismo Gonadal para SQTL tem sido associado a perdas fetais recorrentes durante o terceiro trimestre de gravidez.35 Se a doença é altamente suspeita, a amniocentese após 16 semanas de gestação pode ser útil para estabelecer o diagnóstico, que é facilmente alcançado quando um dos pais é conhecido como portador de uma mutação específica.36
STUDO DE UM PACIENTE COM SÍNDROME QT LONGO PRAZO
História clínica
Uma história familiar e/ou pessoal de morte súbita é de importância crucial para o diagnóstico e estratificação do risco da SQTL. Além disso, fatores precipitantes e o contexto da síncope podem indicar o subtipo da SQTL. Na avaliação inicial de um caso suspeito, o uso de drogas que possam prolongar o intervalo QT deve ser descartado.
Intervalo QT: O que é normal?
O intervalo QT deve ser medido preferencialmente em leads II ou V5,37 onde se comprovou ter maior valor preditivo.38 Este intervalo indica a duração da repolarização ventricular e é medido desde o início da onda Q até o final da onda T. Convencionalmente, a fórmula proposta por Bazett39 é empregada para corrigir a duração do intervalo de acordo com a freqüência cardíaca (QTc=QT/√RR, expressa em segundos). Embora a medida do intervalo QT pareça simples, em um estudo multicêntrico realizado por Viskin et al,40 menos de 40% dos médicos que não são cardiologistas, menos de 50% dos cardiologistas e mais de 80% dos especialistas em arritmia sabiam como mensurá-lo adequadamente. É aconselhável que os médicos realizem medições manuais e não confiem em medições automatizadas, que podem ser úteis para outros intervalos, mas são imprecisas ao calcular o intervalo QT. O QT é um intervalo dinâmico e os limites normais dependem de vários factores. Embora um intervalo QTc de é440 ms em homens e é460 ms em mulheres seja considerado anormal, pode-se encontrar portadores de mutações, bem como indivíduos saudáveis dentro desta faixa (Figura 3). Em famílias com SQTL1, Vincent et al41 demonstraram que nenhum dos casos com genótipo positivo tinha um QTc470 ms. 38 recentemente mostraram que QTc>440 ms é suficiente para detectar pacientes com mutações associadas à SQTL, QTc>470 ms é útil para identificar pacientes em risco de desenvolver sintomas, e QTc>500 ms é encontrado em pacientes sintomáticos submetidos ao tratamento.
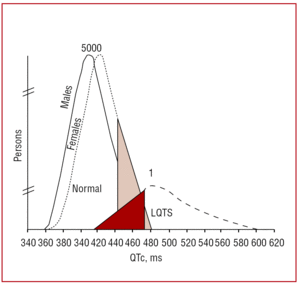
Figure 3. Modelo mostrando a distribuição do intervalo QT (QTc) corrigido pela freqüência cardíaca em pacientes com mutações em KVLQT1, HERG ou SCN5A, e seus familiares não afetados. A curva à esquerda descreve a distribuição dos membros não afetados e a curva à direita, membros afetados.
Outras Alterações Eletrocardiográficas Associadas à Síndrome do QT Longo
Pacientes com SQTL podem apresentar múltiplas alterações da onda T: alternância de polaridade, variações de amplitude, entalhes e aspecto bifásico, entre outras.42 Alternância de onda T (Figura 4A) é definida como uma variação de amplitude, morfologia e polaridade de uma onda T de ritmo sinusal, sem variações no complexo QRS. É um indicador de instabilidade elétrica,43 refletindo a dispersão regional da repolarização ventricular, e ocasionalmente precede a fibrilação ventricular.44
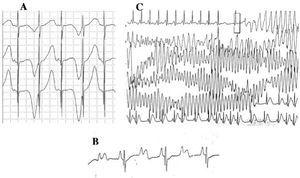
Figure 4. Alterações eletrocardiográficas na síndrome do QT longo. A: Alternância elétrica de onda T. B: Bloqueio atrioventricular 2:1. C: torsade de pointes autolimitada.
Pacientes com SQTL podem progredir com sinais de disfunção do nó sinusal, bradicardia, e/ou pausas.45 Os subtipos SQTL1 e SQTL3, particularmente este último, freqüentemente apresentam bradicardia sinusal,46 enquanto a SQTL4 tem sido associada à disfunção dos nós sinusais.18
Desde a década de 1970-1980, tem sido observada a coexistência de defeitos de condução AV com a SQTL47 (Figura 4B). O bloqueio AV de dois para um é uma manifestação pouco freqüente com um prognóstico ruim que pode estar presente desde a fase fetal na forma de bradicardia persistente. A incidência desta anormalidade tem sido relatada em 4%-5%48 e está associada a alta mortalidade apesar do tratamento com betabloqueadores e/ou marcapassos.49,50 Este fenômeno pode ser explicado por uma longa duração do potencial de ação. Quando o período refratário ventricular é prolongado, o seguinte impulso de atividade sinusal é bloqueado, pois atinge os ventrículos quando estes ainda estão no período refratário. Esta alteração parece ocorrer exclusivamente na SQTL, pois o período refratário ventricular é maior que o do sistema de condução AV.51 A inclinação do complexo QRS é geralmente íngreme e o bloqueio foi localizado na área da infraHis,46,51,52 mas o local do bloqueio pode depender do genótipo. Até o momento, 4 genes foram relacionados ao bloco 2:1 na SQTL: HERG (SQTL2),53,54 SCN5A (SQTL3),52 CACNA1 (SQTL8),26 e SCN4B (SQTL10).55
A arritmia ventricular característica da SQTL é conhecida como torsade de pointes (Figura 4C). Ela se apresenta quando o intervalo QT é prolongado, independentemente da etiologia. É uma taquicardia ventricular polimórfica devido à reentrada, caracterizada eletrocardiograficamente por torção contínua do eixo QRS em torno de uma linha imaginária. É comumente precedida por uma pausa seguida por uma extra-sístole (intervalo RR curto e longo), como mostra a figura.56-58 Pode culminar em fibrilação ventricular e morte súbita. Se isso não ocorrer, o paciente pode apenas experimentar síncope, e se o episódio for breve, pode passar despercebido.
Holter
O estudo Holter fornece uma avaliação completa e dinâmica do intervalo QT. Ocasionalmente são registrados episódios espontâneos de arritmia ventricular assintomática, bem como episódios de disfunção do nó sinusal ou bloqueio AV.
Teste de estresse do exercício
Pacientes com SQTL não podem atingir a freqüência cardíaca máxima esperada calculada de acordo com a idade. Além disso, sob esforço o intervalo QT pode exibir comportamento paradoxal, aumentando ao invés de diminuir.59,60 O padrão eletrocardiográfico durante o teste de estresse do exercício será diferente, dependendo do tipo de SQTL. Os pacientes com SQTL1, além de não alcançarem a freqüência cardíaca máxima calculada para sua idade, freqüentemente apresentam um intervalo QT aumentado, enquanto aqueles com SQTL2 podem alcançar sua freqüência cardíaca esperada e apresentar apenas um leve aumento do intervalo QT ou nenhum aumento.61,62 Em geral, pacientes com SQTL3 têm uma resposta fisiológica ao exercício, ou seja, um encurtamento normal do intervalo QT.63 O teste de estresse também pode ser útil para avaliar a resposta ao tratamento e para estratificar o risco em casos assintomáticos, ou quando há dúvidas quanto aos eventos que levam à arritmia.
Triagem Genética
Nos últimos anos, os estudos genéticos na SQTL têm sido limitados aos laboratórios de pesquisa. No entanto, as informações derivadas desses esforços têm sido extremamente úteis para o tratamento de pacientes, particularmente casos de alto risco. Talvez a principal aplicação da triagem seja no aconselhamento genético, mas também tem implicações importantes no tratamento, que pode ser orientado de acordo com o canal afetado. A localização precisa de uma determinada mutação pode fornecer informações adicionais sobre a evolução do risco. Pacientes com mutações na região transmembrana do KCNQ1 (IKS) têm maior probabilidade de apresentar eventos arrítmicos do que aqueles com mutações na região terminal C64; o mesmo se aplica a pacientes com mutações na região porosa do KCNH2 ou HERG65, em comparação com aqueles com mutações nas regiões N- ou C-terminal.66
O rastreamento inicial talvez se limite aos genes KCNQ1, HERG e SCN5A, que fornecem a possibilidade de encontrar mutações em 65% dos casos. Quando os resultados obtidos são negativos, o rastreamento pode ser estendido para os genes KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 e SCN4B, o que aumentará a possibilidade de resultados positivos em 5% a 10%.
Triagem Genética Pós-morte
É interessante que mutações gênicas que levam à SQTL têm sido encontradas em crianças que experimentaram morte súbita e em casos inexplicáveis de morte súbita em adultos jovens.
Estudos genéticos post-mortem de pacientes com morte súbita e autópsia negativa têm mostrado mutações que levam à SQTL em percentagens variáveis67-69: perto de 10% em crianças e 35% em adultos jovens.70-72 Com base nesses resultados, o estudo ECG de rotina tem sido proposto em todos os recém-nascidos.73,74
Estudo genético post-mortem, também conhecido na literatura como “autópsia molecular”, além das repercussões legais, tem implicações importantes nas famílias que poderiam ser afetadas sem que soubessem disso.
Polimorfismos regulatórios
Vários polimorfismos de ocorrência freqüente têm sido descritos na população da SQTL, distribuídos em quase todos os genes associados a essa condição. Embora estas mudanças aparentemente não sejam patogênicas, algumas podem ter os seguintes efeitos75-78:
1. Gerar susceptibilidade individual para desenvolver arritmia.
2. Favorece o impacto patogênico de outra mudança não-sinônima.
3. Diminui o efeito patogênico de outra mudança não-sinônima.
Este é o caso do polimorfismo K897T no KCNH2 (HERG), que está presente em até 15% da população e não só está ligado à suscetibilidade a certas drogas,79 mas também favorece o efeito patogênico de mutações no mesmo gene.78 Outro exemplo é o polimorfismo S1103Y no gene SCN5A, encontrado principalmente em negros, que tem uma incidência de quase 13% e está associado a um risco aumentado de morte súbita na infância.80
Curiosamente, dois locais de processamento alternativos gerando dois tipos de canais de sódio foram descritos no produto do gene SCN5A, (que codifica o isoform do canal de sódio Nav1,5 em humanos): um com aminoácidos 2016 contendo glutamina na posição 1077 (Q1077), e outro com aminoácidos 2015 sem glutamina (Q1077del). Transcrições destes processos alternativos estão presentes numa proporção 2:1 no mesmo coração humano e vários polimorfismos frequentes terão efeitos diferentes no funcionamento do canal, dependendo se o contexto é Q1077 ou Q1077del. Isto foi inicialmente demonstrado com o polimorfismo H558R do SCN5A, presente em até 30% da população. Quando H558R foi expresso no contexto de Q1077, uma profunda redução na corrente de íons foi observada.81 Um efeito semelhante foi documentado com o polimorfismo de S524Y82. Estes achados forneceram fatores para explicar a gravidade variável da doença, assim como os diferentes fenótipos da mesma mutação observada em algumas famílias.77
Testes farmacológicos com adrenalina
Testes farmacológicos com baixa dose de adrenalina é uma opção segura e útil para desmascarar casos suspeitos de SQTL com uma SQTL limítrofe. É particularmente eficaz para detectar formas assintomáticas de SQTL1, com uma sensibilidade de 92,5%, especificidade de 86%, valor preditivo positivo de 76% e valor preditivo negativo de 96%. Também pode ser útil no diagnóstico da SQTL2, com menor sensibilidade e especificidade. Não é útil para a SQTL3 ou outras formas de SQTL. Em condições normais, a estimulação simpática induz a fosforilação do canal de potássio do IKs, otimizando sua função e dando lugar a encurtamento do potencial de ação. Em pacientes com SQTL, em particular do tipo 1, observa-se uma resposta paradoxal à administração de baixa dose de adrenalina (0,025-0,2 µg/kg/min) que prolonga o intervalo QT para mais de 30 ms83-86.
QT INTERVAL PROLONGATION AND DRUG-INDUCED TORSADE DE POINTES
Uma grande variedade de medicamentos usados em diferentes especialidades médicas pode causar um aumento iatrogênico no intervalo QT. Alguns medicamentos foram retirados do mercado devido a este efeito indesejável (por exemplo, astemizol e cisapride, entre outros; para mais informações, visite www.qtdrugs.org).87,88
A arritmia ventricular secundária a medicamentos não antiarrítmicos ocorre em menos de um de cada 10 000 a 100 000 sujeitos expostos. Considerando que os estudos clínicos incluem entre 2000 e 3000 sujeitos, este evento adverso indesejável e fatal escaparia facilmente à detecção durante a fase clínica de desenvolvimento do fármaco.89 Este ponto tem gerado enorme interesse em aspectos referentes à segurança no estudo e desenvolvimento de novas drogas.
Os fatores relacionados à suscetibilidade individual incluem gênero feminino, hipocalcemia, hipomagnesemia, bradicardia, insuficiência cardíaca, pós-cardioversão, fibrilação atrial, hipertrofia ventricular esquerda, SQTL não detectada, polimorfismos predisponentes e altas concentrações séricas de drogas predisponentes.90
O canal que normalmente interage com drogas é o IKr, codificado pelo gene KCNH2(HERG), por causa de sua estrutura molecular. Outros canais de potássio têm 2 resíduos de prolina angulados em direção ao poro do canal, reduzindo a sua lumínica. Em contraste, o IKr não tem esses resíduos, gera-se um vestíbulo de poro maior e facilita-se a exposição a moléculas grandes. Além disso, tem 2 resíduos aromáticos (tirosina e fenilalanina) que favorecem a ligação com moléculas aromáticas presentes em várias drogas capazes de bloquear o canal.91
Como foi mencionado acima, a penetração da SQTL é incompleta e alguns portadores assintomáticos de mutações podem manifestar arritmias malignas ao receber uma dessas drogas. Além disso, os polimorfismos considerados frequentes na população conferem suscetibilidade individual ao desenvolvimento de torsade de pointes quando algumas drogas são utilizadas. Este é o caso do polimorfismo R1047L, o segundo mais freqüente no KCNH2, que tem sido associado ao desenvolvimento de torsade de pointes com o uso da droga dofetilide.92 Pelo menos 20 polimorfismos do gene KCNH2 têm sido descritos em pessoas saudáveis e seu efeito na suscetibilidade individual ao desenvolvimento de arritmia relacionada a drogas ainda não foi determinado.93 Polimorfismos que conferem suscetibilidade ao desenvolvimento de arritmia ventricular também têm sido documentados no canal de sódio Na1,5. É o caso do polimorfismo H558R, presente em até 30% da população, ou S1103Y, freqüente em negros80,81,90,94,95; sua implicação na susceptibilidade induzida por drogas não foi investigada.
LONG QT SÍNDROME E PREGNÂNCIA
O aconselhamento genético é importante na SQTL, mas em termos gerais não há contra-indicação para gravidez em mulheres portadoras, embora cada caso seja diferente e deva ser avaliado individualmente no contexto apropriado.
Foi observado que o risco de apresentar arritmia ventricular maligna diminui com a gravidez. Em contraste, maior vulnerabilidade para apresentar arritmia maligna tem sido relatada nos primeiros 9 meses após o parto, particularmente em pacientes com SQTL2. Esse risco diminui consideravelmente com a terapia com beta-bloqueador.96
ESTRATIFICAÇÃO DO RISCO
A evolução da SQTL varia e é influenciada pela duração do intervalo QTc, fatores ambientais, idade, genótipo e resposta ao tratamento.97,98 A arritmia ventricular é mais frequente na SQTL1 e SQTL2, mas é mais grave na SQTL3.99 Como foi mencionado acima, as mulheres são especialmente suscetíveis à arritmia maligna durante o período pós-parto.14
Síndrome do QT Longo deve ser considerada de alto risco quando associada com o seguinte:
1. Surdez congênita (síndrome de Jervell-Lange-Nielsen).
2. Síncope recorrente devido a taquiarritmia ventricular maligna.
3. História familiar de morte súbita.
4. QTc>500 ms.
5. 2:1 bloqueio atrioventricular.
6. O estudo de Priori et al97 realizado em 647 pacientes mostrou que a probabilidade de apresentar um evento maior (síncope, parada cardíaca, morte súbita) antes dos 40 anos é alta (>50%) quando QTc é >500 ms na SQTL1, SQTL2, e em homens com SQTL3. Recentemente, uma análise do registro internacional da SQTL foi relatada. O risco de morte súbita foi analisado em 2772 adolescentes com a doença, e 3 fatores associados com maior risco nesta população foram identificados: QTc>530 ms, história de síncope nos últimos 10 anos, e sexo; meninos de 10 a 12 anos de idade tinham maior risco do que meninas, mas na faixa etária de 13 a 20 anos, o risco era comparável.100
TREATAMENTO
Pacientes sintomáticos que não recebem tratamento têm uma taxa de mortalidade anual de 20% e mortalidade de 10 anos de 50% após um primeiro evento de arritmia ventricular. Embora seja claro que o tratamento deve ser estabelecido quando há sintomas, a abordagem a ser usada em pacientes assintomáticos ainda está em debate. Tem sido documentado que a parada cardíaca pode ser a primeira manifestação da doença em 9% dos pacientes,48 e que 12% dos pacientes assintomáticos desenvolverão sintomas e poderão sofrer morte súbita. O tratamento inicial com beta-bloqueadores deve ser iniciado em todos os pacientes com SQTL. A restrição do exercício é recomendável, mas os marcadores de risco clínicos e eletrocardiográficos são uma base útil para a tomada de decisões. É importante informar os pacientes sobre o risco do uso de várias drogas que podem prolongar o intervalo QT e favorecer o desenvolvimento de arritmia ventricular, como mencionado acima. O diagnóstico genético, além de permitir o aconselhamento familiar adequado relacionado à doença, é uma ajuda para avaliar o prognóstico e orientar o tratamento específico.
Beta-Blockers
Beta-bloqueadores são o tratamento de primeira linha da SQTL e todos os pacientes devem recebê-los como a terapia inicial.101 Eles proporcionam uma redução no risco de eventos cardiovasculares de até 64%100 e são particularmente eficazes em pacientes com mutações do canal IKs (LQTS1),102 que são regulados em grande parte pelo sistema simpático. Os beta-bloqueadores não modificam o intervalo QT, mas sim sua dispersão.103 Embora esses medicamentos diminuam a incidência de eventos,104,105 foi demonstrado que 10% dos pacientes com SQTL1, 23% com SQTL2 e 32% com SQTL3 terão sintomas cardiovasculares apesar do tratamento.106 Os pacientes com SQTL3, em particular, não parecem obter benefícios importantes; de fato, este grupo de medicamentos deve ser usado com cautela nestes pacientes, pois os episódios de arritmia ventricular na SQTL3 são mais comuns quando a freqüência cardíaca está baixa. Em termos gerais, 32% dos pacientes sintomáticos terão sintomas recorrentes nos primeiros 5 anos antes de iniciar o tratamento com beta-bloqueador, e 14% dos pacientes resgatados de um episódio de morte súbita apresentarão outro evento similar dentro de 5 anos se receberem apenas esta terapia.107 Vários beta-bloqueadores têm sido utilizados no tratamento da SQTL, principalmente nadolol (0,5-1 mg/kg/dia), propranolol (2-4 mg/kg/dia), metoprolol (0,5-1 mg/kg/dia) e atenololol (0,5-1 mg/kg/dia). O atenolol pode não ser benéfico na SQTL, entretanto, foi notificado que pelo menos 75% dos pacientes que não responderam à terapia com beta-bloqueador estavam recebendo atenolol, embora este achado possa estar relacionado ao uso de doses subótimas.104 O teste de exercício é útil para estabelecer a dose apropriada. A freqüência cardíaca máxima não deve exceder 130 batimentos/min durante o tratamento.
Bloqueadores do canal de sódio
Mutações do canal de sódio que causam LQTS3 produzem inativação defeituosa do canal; o bloqueio do canal de sódio provou ser útil nesses pacientes. Estudos feitos com flecainida documentaram melhorias na freqüência cardíaca, alterações da onda T e intervalo QT.108 Mexiletine também tem sido relatado para melhorar os marcadores de risco eletrocardiográfico.63.109.110 Estudos in vitro com ranolazina têm mostrado diminuições nos efeitos deletérios das mutações relatadas em humanos.111 Embora os resultados sejam encorajadores, deve-se ter em mente que não há estudos de longo prazo avaliando esta terapia, e nenhum achado relatado de grandes séries. Os bloqueadores dos canais de sódio não devem ser administrados se não houver diagnóstico genético confirmado.
Suplementação de Potássio e Drogas que Aumentam sua Disponibilidade
Suplementação de Potássio e/ou drogas que economizam potássio, como a espironolactona, encurtam o intervalo QTc em 24% dos casos.112.113 Medicamentos que favorecem a abertura dos canais de potássio, tais como aprikalim, levcromakalim, nicorandil e pinacidil, demonstraram ser úteis no tratamento da SQTL. Os subtipos em que eles são particularmente benéficos são SQTL1 e SQTL2.114
Marcapassos e desfibriladores
Estimulação com marcapasso tem sido usada em pacientes com arritmia pause-dependente.115,116 Pacientes com SQTL3 geralmente se beneficiam mais deste tratamento porque a prevalência de bradicardia é maior neste grupo. A estimulação DDD é indicada em pacientes com arritmia pause-dependente ou bloqueio AV de alto grau 2:1. Frequências programadas abaixo de 70 batimentos/min117 não são úteis para prevenir a arritmia ventricular. Recomenda-se programar o sensor para resposta rápida, pois esses pacientes geralmente têm aceleração inadequada da freqüência cardíaca em resposta ao exercício. Todas as funções que implicam a presença de pausas devem ser desligadas, como a histerese e a função noturna. A PARP (período pós-ventricular atrial refratário) deve ser o mais curto possível. A função de regulação de freqüência deve estar ligada para evitar pausas pós-extrasistólicas. Deve-se lembrar que o excesso de onda T e as falhas de captura também podem dar origem a pausas. O uso combinado de cardioversor desfibrilador implantável (CDI) e betabloqueadores diminui substancialmente a incidência de morte súbita.118-120 A indicação destas medidas é clara nos casos de alto risco.121 A programação do dispositivo varia de acordo com as necessidades de cada paciente, mas, geralmente, a administração do tratamento em eventos assintomáticos e autolimitados deve ser evitada; para este fim, é indicado um tempo de detecção de 15 s. A tempestade arrítmica é uma complicação da terapia AID. Cerca de 15% dos pacientes podem experimentar esta complicação, que se deve, em boa parte, ao aumento do tom simpático após o choque do CDI.118 Este problema pode ser gerenciado através do aumento da dose do beta-bloqueador. Se esta medida não for útil, a ressecção dos gânglios da cadeia simpática deve ser considerada.
Simpatectomia esquerda
Em 1971, a gangliectomia simpática foi introduzida como uma opção terapêutica útil nestes pacientes.122 Em 1991, Schwartz et al123 publicaram a primeira série de 85 pacientes com má resposta ao tratamento com beta-bloqueador, nos quais uma estelectomia esquerda foi realizada com resultados encorajadores: uma taxa de sobrevida de 5 anos de 94%. Atualmente, essa opção terapêutica é oferecida aos pacientes de alto risco que persistem com síncope apesar do tratamento com betabloqueador e/ou implante de marcapasso, e àqueles que experimentam choques freqüentes do desfibrilador implantado. O procedimento consiste na ressecção da porção inferior do gânglio estrelado e dos gânglios torácicos esquerdos T2 a T4 da cadeia simpática, uma vez que a estelectomia simples esquerda não se mostrou suficientemente eficaz. A toracoscopia microinvasiva124.125 tem sido utilizada com bons resultados. A maior série de pacientes tratados com este método foi relatada recentemente e mostrou uma redução significativa no número de episódios de síncope ou mortes súbitas, assim como uma taxa de sobrevida de 5 anos de 95%. Em pacientes com síncope prévia, a sobrevida em 5 anos foi de 97%, com 11% de possibilidade de recidiva, que, em sua maioria, consistiu de um único evento sincopal. Houve também uma redução significativa no segmento QT após a simpatectomia esquerda. Apesar destes resultados favoráveis, a prevenção de morte súbita não está completa, mas foi reduzida para 3%. Em pacientes com CDI que se submeteram a cirurgia por causa de choques desfibriladores múltiplos, o número médio de eventos diminuiu de 25 para 0, uma redução de 95%. Um efeito benéfico foi confirmado na SQTL1. Os benefícios são provavelmente menores em pacientes com SQTL2, e na SQTL3, sua eficácia não foi comprovada.126
Ablação
Foi relatado que a ablação da extra-sístole, que em alguns casos inicia a arritmia ventricular, pode ser realizada com a redução da incidência de episódios.127 Entretanto, não há estudos a longo prazo com um número apropriado de pacientes para justificar o uso rotineiro desta técnica.
Veja editorial nas páginas 675-82
ABREVIATIONS
AV: atrioventricular
AID: desfibrilador implantável automático
ECG: eletrocardiograma
QTc: freqüência cardíaca corrigida QT
ATS: Síndrome de Andersen-Tawil
SQTL: síndrome do QT longo
Dr. Medeiros recebe apoio econômico da CONACyT e FUNSALUD.