INTRODUCERE
Sindromul QT lung (LQTS) este caracterizat de o repolarizare ventriculară grav alterată, care determină prelungirea intervalului QT pe electrocardiogramă (ECG). Afecțiunea predispune pacienții la aritmie ventriculară malignă (torsade de pointes) și moarte subită. Descrierea clinică și electrocardiografică a sindromului QT lung a fost raportată în 1957 de către Anton Jervell și Fred Lange Nielsen,1 care și-au publicat studiile asupra unei familii de părinți neconsangvinizați cu 6 copii. Patru dintre copii aveau surditate congenitală și episoade sincopale, iar 3 au prezentat moarte subită. Studiul ECG al acestor pacienți a arătat un interval QT neobișnuit de lung. Ambii părinți au fost asimptomatici, au avut un ECG normal și nu au prezentat probleme de auz. În 1964, Romano și Ward au raportat independent un sindrom cardiac caracterizat prin sincope recurente, antecedente familiale de moarte subită și prelungirea intervalului QT fără surditate neuronală.2 Studiile genetice ulterioare au arătat că sindromul descris de Jervell și Lange Nielsen, care este însoțit de surditate neuronală congenitală, corespunde unor mutații homozigote, cu un fenotip sever și risc ridicat de moarte subită. Afecțiunea cunoscută sub numele de sindrom Romano-Ward corespunde, în general, mutațiilor heterozigote, pacienții nu prezintă alterări ale auzului, iar severitatea bolii variază considerabil. Aproape o jumătate de secol mai târziu, în 1995,3,4 au fost descrise principalele gene asociate cu LQTS, iar boala a fost recunoscută ca fiind o tulburare a canalelor ionice cardiace. A fost prima canalopatie cardiacă descrisă și este probabil cea mai investigată tulburare aritmogenă a canalelor ionice aritmogene până în prezent. Tabloul clinic variază foarte mult: pacientul poate fi asimptomatic sau poate prezenta sincope recurente, convulsii sau moarte subită ca primă manifestare a bolii. Inițial, LQTS a fost considerat un sindrom rar și, de fapt, prezentarea severă a bolii este sporadică. Cu toate acestea, incidența mutațiilor înrudite este estimată la 1/3000-5000 de cazuri,5 32% dintre purtătorii asimptomatici pot avea un interval QT corectat pentru frecvența cardiacă (QTc) în limite normale, boala este transmisă la 50% dintre descendenții lor, aceștia sunt mai susceptibili de a dezvolta aritmii în comparație cu populația generală și până la 20% pot deveni simptomatici.6
Sindromul QT lung prezintă o mare eterogenitate genetică. Mai mult de 500 de mutații distribuite în 10 gene au fost descrise în această afecțiune: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 și SCN4B. În ciuda progreselor din acest domeniu, nu se poate stabili un diagnostic genetic la 25%-30% dintre pacienți.7,8 Prezentarea bolii este în principal monogenică6; varietățile poligene sau compuse au de obicei un fenotip mai sever. Penetranța, adică pacienții care au mutația și manifestă fenotipul, variază între 25% și 90%.9 Mai puțin frecvent, pot exista variații în expresivitatea bolii, cu mai multe fenotipuri rezultate din aceeași mutație. Studiile de genetică moleculară dezvoltate în ultimii 11 ani au adus corelații genotip-fenotip importante, care au contribuit la orientarea abordării terapeutice. În plus, în cadrul studiilor care au investigat polimorfismele nesinonime frecvente în această populație, au fost făcute observații interesante privind susceptibilitatea individuală de a dezvolta aritmie, aspect care a stârnit un interes considerabil, în special în domeniul farmacogenomicii.
CLASIFICAREA SINDROMULUI QT LUNG
Concepte generale
Clasificarea LQTS folosită în trecut se baza pe prezentarea homozigotă sau heterozigotă a bolii, care dă naștere la sindromul Jervell-Lange-Nielsen (cu surditate) și, respectiv, sindromul Romano-Ward (fără surditate). Clasificarea actuală pune accentul pe constatările genetice, așa cum este ilustrat în tabelul 1. Cele 3 gene principale asociate cu boala au fost descrise în 1995-1996. Aceste gene, care codifică unitățile formatoare de pori ale canalelor de potasiu IKs și IKr, precum și canalul de sodiu Nav1.5, reprezintă aproape 65% din cazuri. Deși în anii următori alte șapte gene au fost incluse în listă, acestea reprezintă doar 5% din cazuri.
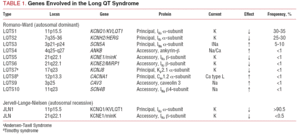
Canalele de ioni sunt proteine transmembranare care transportă ioni prin membrana celulară. Canalele implicate în LQTS sunt selective sau specializate în transportul unui singur ion și sunt voltaj-dependente, adică activarea lor are loc la un voltaj intracelular specific, care variază în funcție de subtipul canalului. Fenomenele electrice și contractile care au loc în cardiomiocite sunt controlate de aceste structuri. Canalele ionice formează complexe macromoleculare alcătuite dintr-o unitate principală care formează porul canalului și proteine auxiliare care îl reglează (figura 1). Disfuncția canalului observată în LQTS poate să apară la aceste două locații: proteina principală sau proteinele de reglare (tabelul 1). Implicarea unității care formează porul, cunoscută sub numele de alfa, generează cele mai frecvente trei subtipuri de LQTS: LQTS1 (care afectează canalul de potasiu IKs), LQTS2 (care afectează canalul de potasiu IKr) și LQTS3 (care afectează canalul de sodiu). Deoarece acestea sunt cele mai frecvente subtipuri, ele sunt cele mai bine caracterizate clinic și genetic. Corelațiile fenotip-genotip în aceste trei forme principale sunt descrise în figura 2. În prezent, sindromul Jervell-Lange-Nielsen corespunde varietăților LQTS 1 și 5. În mod caracteristic, acești pacienți au surditate congenitală și mutații homozigote sau heterozigote compuse care afectează curentul IKs. Sindromul Romano-Ward include varietățile de la LQTS 1 la 10 și nu implică surditate.
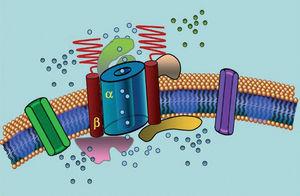
Figura 1. Reprezentarea schematică a complexului macromolecular. Canalele ionice sunt proteine transmembranare (α) reglementate de diverse proteine; una dintre ele este așa-numita subunitate β.
Sindromul QT lung de tip 1 (LQTS1)
Pacienții cu LQTS1 prezintă de obicei episoade de aritmie ventriculară atunci când fac exerciții fizice sau când sunt supuși unui stimul simpatic (68%).10 Înotul a fost descris ca un sport care declanșează aritmia în LQTS1.11 Penetranța este de aproape 62% în acest subtip. Unda T la acești pacienți are adesea o bază largă și o durată foarte prelungită12,13 (figura 2). Este cel mai frecvent subtip și explică 30%-35% din cazuri. Gena afectată, KvLQT1 (sau KCNQ1), este localizată pe cromozomul 11 (11p15.5) și codifică pentru subunitatea α a canalului de potasiu IKs. Potențialul de acțiune este prelungit de o reducere a curentului K+ de ieșire în timpul fazei 3 a potențialului de acțiune.
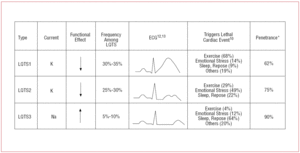
Figura 2. Corelația genotip-fenotip în cele mai frecvente sindroame QT lung. *Se referă la cazurile care au mutația și manifestă fenotipul.
Sindromul QT lung de tip 2 (LQTS2)
Pacienții cu LQTS2 tind să prezinte aritmie ventriculară ca răspuns la stres emoțional (49%) sau la stimuli auditivi bruște (de exemplu, un ceas deșteptător), și mai puțin frecvent în timpul somnului (22%) sau al exercițiilor fizice (29%).10 Femeile în perioada postpartum sunt deosebit de susceptibile.14 Penetranța estimată este de 79%; prin urmare, până la 20% din cazuri pot avea un ECG nediagnostic. Unda T în LQTS2 este de obicei de amplitudine mică și bifidă, cu crestături12,13 (figura 2). Gena afectată este KCNH2 sau HERG, localizată pe cromozomul 7 (7q35-36), care codifică pentru subunitatea α a canalului de potasiu IKr și reprezintă 25%-30% din cazuri. Disfuncția acestui canal scade curentul K+ de ieșire în timpul fazei 3 a potențialului de acțiune, prelungind durata acestuia.
Sindromul QT lung tip 3 (LQTS3)
Pacienții cu LQTS3 au un risc mai mare de a prezenta aritmii maligne în timpul repausului (somn) sau bradicardie.15 Penetranța mutației genei SCN5A este de aproape 90%. ECG-ul în LQTS3 prezintă de obicei o undă T întârziată, ascuțită și permite observarea clară a prelungirii segmentului ST12,13 (figura 2). Acești pacienți au de obicei mai puține simptome decât cei cu LQTS1 sau LQTS2, dar evenimentele sunt în mod caracteristic mai letale.
Gena afectată în LQTS3 este SCN5A, care codifică pentru canalul de sodiu Nav1.5 α-subunitatea (figura 1), localizată pe cromozomul 3 (3p21-24); este cauza bolii în 5%-10% din cazuri. Inactivarea defectuoasă a canalului permite o intrare susținută de Na+ în timpul fazei 2 a potențialului de acțiune, prelungind durata acestuia.
Sindromul QT lung de tip 4 (LQTS4)
Tipul 4 este o varietate rară de LQTS, reprezentând aproape 1% din cazuri. Este o formă atipică care produce un spectru larg de aritmii, inclusiv tahicardie ventriculară polimorfă catecolaminergică, fibrilație atrială, alterări ale conducerii intraventriculare, disfuncție a nodului sinusal și bradicardie6-18; în plus, QTc poate fi în limite normale la mulți pacienți. Gena afectată este ANKB, localizată pe cromozomul 4 (4q25-27), care codifică pentru sinteza ankyrin-β, o proteină structurală care leagă proteinele membranare ale cardiomiocitelor de proteinele citoscheletale. Aceste proteine sunt pompa Na/K ATPază, schimbătorul Na/Ca și receptorul de inositol trifosfat (InsP3R). Mutațiile care cauzează o pierdere a funcției anchirinei-β conduc la creșteri ale concentrației intracelulare de calciu și la modificări ale expresiei ATPazei N/K și a schimbătorului Na/Ca. Concentrația crescută de calciu dă naștere la post-depolarizări timpurii și întârziate. Astfel, aritmiile ventriculare observate în cazul mutațiilor genei ankyrin-β se datorează unor depolarizări spontane, de obicei ca răspuns la stimularea catecolaminergică.
Sindromul QT lung tip 5 (LQTS5)
Tipul 5 își are originea în modificări ale secvenței genei KCNE1 situată pe cromozomul 21 (21q22.1p22.)19 KCNE1 codifică pentru sinteza subunității β a canalului IKs, cunoscută și subunitatea minK, care reglează canalul IKs. Acest tip reprezintă mai puțin de 1% din cazuri.
Sindromul QT lung tip 6 (LQTS6)
Gena afectată în tipul 6 este KCNE2, localizată pe cromozomul 21 (21q22.1).20 Această genă codifică pentru subunitatea β a canalului de potasiu, cunoscută și subunitatea MiRP1, și reglează canalul IKr. Mai puțin de 1% din cazuri sunt de tip 6.
Sindromul QT lung de tip 7 sau Sindromul Andersen-Tawil (LQTS7)
Dezvăluirile dismorfice și alterările electrocardiografice observate în acest sindrom au fost descrise pentru prima dată în 1971 de către Dr. Andersen21 și revizuite în 1994 de către Dr. Tawil,22 dar descrierea genetică/moleculară nu a fost raportată până în 2001.23 Cunoscută în prezent sub numele de sindromul Andersen-Tawil (ATS), această afecțiune este o alterare autozomal dominantă caracterizată prin paralizie periodică, dezvoltare anormală a scheletului, aritmie ventriculară de tipul celor care implică extrasistole ventriculare frecvente și o predispoziție deosebită de a dezvolta fibrilație ventriculară, în special la femei. Alterările descrise la ATS includ extrasistole ventriculare (41%), tahicardie ventriculară polimorfă polimorfă nesusținută (23%), tahicardie ventriculară bidirecțională (68%) și torsade de pointes (3%).24 Unele dintre caracteristicile dismorfice observate includ statura scurtă, scolioza, clinodactilia, hipertelorismul, implantarea joasă a urechilor, micrognatia și o frunte lată. Expresia bolii variază, fapt care complică diagnosticul precoce.23,25 Mutațiile în gena KCNJ2 situată în cromozomul 17 (17q23), care codifică pentru sinteza canalului de potasiu rectificator Kir 2.1, reprezintă 70% din cazuri. Acest canal participă la faza 4 a potențialului de acțiune. Mai mulți autori pun sub semnul întrebării includerea acestei gene în cadrul grupului cauzal LQTS, deoarece intervalul QTc este doar ușor prelungit în acest sindrom sau chiar normal, dar unda U este de obicei proeminentă, ceea ce a dus la supraestimarea intervalului QT. Cititorul va constata că unii autori sugerează că mutațiile KCNJ2 generează ATS1 și nu LQTS7.24
Sindromul QT lung de tip 8 (LQTS8)
Tipul 8 apare în urma unor mutații în gena CACNA1 situată pe cromozomul 12 (12p13.3), care codifică canalul de calciu de tip L Cav1.2. Aceasta determină sindromul Timothy26 , o afecțiune caracterizată prin malformații cardiace, deficiență imunologică intermitentă, hipoglicemie, alterări cognitive, inclusiv autism, fuziune interdigitală și QT prelungit, care duce la aritmie cardiacă și moarte subită27. Mai puțin de 0,5% din cazuri sunt de tip 8.
Sindromul QT lung de tip 9 (LQTS9)
Această varietate de LQTS se dezvoltă prin mutații în gena CAV3, situată pe cromozomul 3 (3p25), care codifică pentru sinteza caveolinei 3. Caveolina este o invaginație a membranei plasmatice implicată în endocitoză, homeostazia lipidelor și transducția semnalelor. O componentă importantă a acestei structuri este caveolina, care are 3 subtipuri cunoscute; subtipul 3 este specific pentru mușchiul scheletic și cardiac. Unele canale ionice sunt co-localizate în caveolă, inclusiv o izoformă cardiacă a canalului de sodiu Nav1.5. Mai multe mutații în această proteină au fost descrise recent. Acestea modifică proprietățile biofizice ale canalului de sodiu Nav1.5 in vitro, generând un fenotip similar cu cel observat în LQTS3.28 Mai puțin de 1% din cazuri sunt atribuite acestei cauze.
Sindromul QT lung tip 10 (LQTS10)
Tipul 10 a fost descris într-un caz foarte sever, cu QTc >600 ms, bradicardie fetală și bloc atrioventricular (AV) 2:1. Este rezultatul unor mutații în gena SCN4B, localizată pe cromozomul 11 (11q23), care codifică pentru subunitatea β4 a canalului de sodiu. Au fost descrise patru subtipuri diferite de subunități β, care interacționează și reglează diferitele izoforme ale canalului de sodiu; cu toate acestea, până în prezent, doar subtipul 4 a fost asociat cu aritmogeneza.29 Incidența mutațiilor acestei gene nu a fost examinată, dar este estimată la
Mutații ale varietății Jervell-Lange-Nielsen
Această formă severă de LQTS este cauzată de mutații homozigote30 sau heterozigote compuse ale genelor KCNQ1, și/sau KCNE1, care codifică pentru curentul IKs; adică, o varietate foarte severă a formelor LQTS1 sau LQTS5. Această afecțiune este asociată în mod caracteristic cu surditatea congenitală. Pacienții au, de obicei, un QTc>500 ms și sincope recurente și prezintă un risc ridicat de moarte subită. Părinții pacienților cu această varietate sunt de obicei heterozigoți și au o boală mai puțin severă sau nu prezintă simptome.31
DIAGNOSTICUL SINDROMULUI QT LUNG
Scorul Schwartz
În 1985, Schwartz et al32 au publicat criteriile de diagnosticare a SQTL, care au fost modificate în 1993 și care conțin orientări importante pentru evaluarea inițială a cazurilor potențiale. Acest sistem utilizează un scor de la 1 la 9 pe baza istoricului familial și a constatărilor clinice și electrocardiografice. Probabilitatea de boală este scăzută la un scor ≥1, intermediară la 2-3 și ridicată la ≥4 (tabelul 2).

Diagnosticul prenatal al sindromului QT lung
Bradicardia fetală poate fi una dintre primele manifestări clinice ale SQTL. Seriile retrospective au arătat că până la 70% dintre pacienții diagnosticați cu SQTL în copilărie au un istoric de bradicardie, de obicei însoțit de hidrops fetal.33 Evaluarea repolarizării cardiace fetale între săptămânile 14 și 39 este utilă pentru diagnosticarea precoce a SQTL.34
Mozaicismul gonadal pentru SQTL a fost asociat cu pierderi fetale recurente în timpul celui de-al treilea trimestru de sarcină.35 În cazul în care boala este foarte suspectată, amniocenteza după 16 săptămâni de gestație poate fi utilă pentru stabilirea diagnosticului, care este ușor de atins atunci când se știe că unul dintre părinți este purtător al unei mutații specifice.36
STUDIUL UNUI PACIENT CU SINDROMUL QT LUNG
Anamneza clinică
Anamneza familială și/sau personală a morții subite este de o importanță crucială atât pentru diagnosticul cât și pentru stratificarea riscului de SQTL. În plus, factorii precipitanți și contextul sincopei pot indica subtipul LQTS. În evaluarea inițială a unui caz suspect, trebuie exclusă utilizarea medicamentelor care pot prelungi intervalul QT.
Intervalul QT: Ce este normal?
Intervalul QT trebuie măsurat preferențial în derivațiile II sau V5,37 unde s-a dovedit că are o valoare predictivă mai mare.38 Acest interval indică durata repolarizării ventriculare și se măsoară de la începutul undei Q până la sfârșitul undei T. În mod convențional, se utilizează formula propusă de Bazett39 pentru a corecta durata intervalului în funcție de frecvența cardiacă (QTc=QT/√RR, exprimată în secunde). Deși măsurarea intervalului QT pare simplă, într-un studiu multicentric realizat de Viskin et al40, mai puțin de 40% dintre medicii, alții decât cardiologii, mai puțin de 50% dintre cardiologi și mai mult de 80% dintre specialiștii în aritmii știau cum să îl măsoare corect. Este recomandabil ca medicii să efectueze măsurători manuale și să nu se încreadă în măsurătorile automate, care pot fi utile pentru alte intervale, dar sunt imprecise la calcularea intervalului QT. QT este un interval dinamic, iar limitele normale depind de mai mulți factori. Deși un interval QTc de é440 ms la bărbați și é460 ms la femei este considerat anormal, se pot găsi atât purtători de mutații, cât și persoane sănătoase în acest interval (figura 3). În familiile cu LQTS1, Vincent et al41 au demonstrat că niciunul dintre cazurile cu un genotip pozitiv nu a avut un QTc470 ms. Monnig et al38 au arătat recent că QTc>440 ms este suficient pentru a detecta pacienții cu mutații asociate LQTS, QTc>470 ms este util pentru a identifica pacienții cu risc de a dezvolta simptome, iar QTc>500 ms se regăsește la pacienții simptomatici aflați sub tratament.
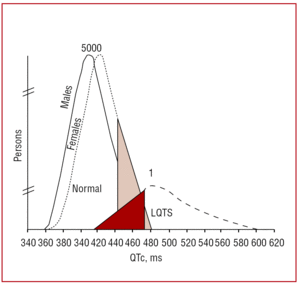
Figura 3. Modelul care arată distribuția intervalului QT corectat în funcție de frecvența cardiacă (QTc) la pacienții cu mutații în KVLQT1, HERG sau SCN5A și la membrii familiilor lor neafectați. Curba din stânga descrie distribuția membrilor neafectați, iar curba din dreapta, membrii afectați.
Alte modificări electrocardiografice asociate cu sindromul QT lung
Pacienții cu LQTS pot prezenta multiple modificări ale undei T: alternanță de polaritate, variații de amplitudine, crestături și un aspect bifazic, printre altele.42 Alternanța undei T (figura 4A) este definită ca o variație de la bătaie la bătaie a amplitudinii, morfologiei și polarității unei unde T în ritm sinusal, fără variații ale complexului QRS. Este un indicator al instabilității electrice,43 reflectând dispersia regională a repolarizării ventriculare și, ocazional, precede fibrilația ventriculară.44
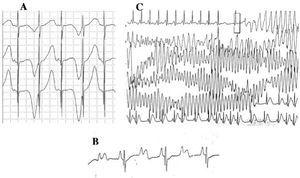
Figura 4. Modificări electrocardiografice în sindromul QT lung. A: Alternanță electrică a undei T. B: bloc atrioventricular 2:1. C: torsade de pointes autolimitată.
Pacienții cu SQTL pot evolua cu semne de disfuncție a nodului sinusal, bradicardie și/sau pauze. 45 Subtipurile LQTS1 și LQTS3, în special cel din urmă, prezintă adesea bradicardie sinusală,46 în timp ce LQTS4 a fost asociat cu disfuncția nodului sinusal.18
Începând cu deceniul 1970-1980, a fost observată coexistența defectelor de conducere AV cu LQTS47 (figura 4B). Blocul AV doi la unu este o manifestare puțin frecventă, cu un prognostic nefavorabil, care poate fi prezentă încă din stadiul fetal sub forma unei bradicardii persistente. Incidența acestei anomalii a fost raportată la 4%-5%48 și este asociată cu o mortalitate ridicată în ciuda tratamentului cu beta-blocante și/sau stimulatoare cardiace.49,50 Acest fenomen poate fi explicat printr-o durată prelungită a potențialului de acțiune. Atunci când perioada refractară ventriculară este prelungită, impulsul următor al activității sinusale este blocat, deoarece ajunge la ventricule când acestea se află încă în perioada refractară. Această alterare pare să apară exclusiv în SQTL, deoarece perioada refractară ventriculară este mai mare decât cea a sistemului de conducere AV.51 Panta complexului QRS este de obicei abruptă, iar blocajul a fost localizat în zona infraHis,46,51,52 dar locul blocului poate depinde de genotip. Până în prezent, 4 gene au fost legate de blocajul 2:1 în LQTS: HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 și SCN4B (LQTS10).55
Aritmia ventriculară caracteristică a LQTS este cunoscută sub numele de torsade de pointes (figura 4C). Aceasta se prezintă atunci când intervalul QT este prelungit, indiferent de etiologie. Este o tahicardie ventriculară polimorfă datorată reintrării, caracterizată electrocardiografic prin răsucirea continuă a axei QRS în jurul unei linii imaginare. Este de obicei precedată de o pauză urmată de o extrasistolă (interval RR scurt-lung-scurt), așa cum se arată în figură.56-58 Poate culmina cu fibrilație ventriculară și moarte subită. Dacă aceasta nu se produce, pacientul poate prezenta doar o sincopă, iar dacă episodul este scurt, poate trece neobservat.
Studiul Holter
Studiul Holter oferă o evaluare completă și dinamică a intervalului QT. Ocazional sunt înregistrate episoade spontane de aritmie ventriculară asimptomatică, precum și episoade de disfuncție a nodului sinusal sau bloc AV.
Test de efort
Pacienții cu SQTL nu pot atinge frecvența cardiacă maximă așteptată calculată în funcție de vârstă. În plus, în condiții de efort, intervalul QT poate prezenta un comportament paradoxal, prin creștere în loc să scadă.59,60 Modelul electrocardiografic în timpul testului de efort va fi diferit în funcție de tipul de SQTL. Pacienții cu LQTS1, pe lângă faptul că nu ating frecvența cardiacă maximă calculată pentru vârsta lor, prezintă frecvent o creștere a intervalului QT, în timp ce cei cu LQTS2 pot atinge frecvența cardiacă așteptată și prezintă doar o creștere ușoară a intervalului QT sau deloc.61,62 În general, pacienții cu LQTS3 au un răspuns fiziologic la efort, adică o scurtare normală a intervalului QT.63 Testul de efort poate fi, de asemenea, util pentru evaluarea răspunsului la tratament și pentru stratificarea riscului în cazurile asimptomatice sau atunci când există îndoieli cu privire la evenimentele care conduc la aritmie.
Screening genetic
În ultimii ani, studiile genetice în LQTS au fost limitate la laboratoarele de cercetare. Cu toate acestea, informațiile derivate din aceste eforturi au fost extrem de utile pentru tratarea pacienților, în special a cazurilor cu risc ridicat. Poate că principala aplicație a screening-ului este în consilierea genetică, dar are, de asemenea, implicații importante în tratament, care poate fi orientat în funcție de canalul afectat. Localizarea precisă a unei anumite mutații poate oferi informații suplimentare cu privire la evoluția riscului. Pacienții cu mutații în regiunea transmembranară a KCNQ1 (IKS) au o probabilitate mai mare de a prezenta evenimente aritmice decât cei cu mutații în regiunea C-terminală64; același lucru este valabil și în cazul pacienților cu mutații în regiunea porilor KCNH2 sau HERG65 în comparație cu cei cu mutații în regiunile N- sau C-terminală.66
Screeningul inițial poate fi poate limitat la genele KCNQ1, HERG și SCN5A, care oferă posibilitatea de a întâlni mutații în 65% din cazuri. Atunci când rezultatele obținute sunt negative, screeningul poate fi extins la genele KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 și SCN4B, ceea ce va crește posibilitatea de a obține rezultate pozitive cu 5% până la 10%.
Screening genetic postmortem
Este interesant faptul că mutațiile genetice care duc la SQTL au fost găsite la copii care au suferit moarte subită și în cazuri inexplicabile de moarte subită la adulți tineri.
Studiile genetice postmortem ale pacienților cu moarte subită și autopsie negativă au evidențiat mutații care conduc la LQTS în procente variabile67-69: aproape 10% la copii și 35% la adulții tineri70-72. Pe baza acestor rezultate, a fost propus studiul ECG de rutină la toți nou-născuții.73,74
Studiul genetic postmortem, cunoscut în literatura de specialitate și sub numele de „autopsie moleculară”, pe lângă repercusiunile legale, are implicații importante în familiile care ar putea fi afectate fără ca acestea să știe.
Polimorfisme reglatoare
În populația cu SQTL au fost descrise mai multe polimorfisme care apar frecvent, distribuite în aproape toate genele asociate cu această afecțiune. Deși aceste modificări nu sunt aparent patogene, unele dintre ele pot avea următoarele efecte75-78:
1. Generează susceptibilitate individuală de a dezvolta aritmie.
2. 2. Favorizează impactul patogen al unei alte modificări nesinonime.
3. Diminuează efectul patogen al unei alte modificări nesinonime.
Acesta este cazul polimorfismului K897T în KCNH2 (HERG), care este prezent la până la 15% din populație și care nu numai că este legat de susceptibilitatea la anumite medicamente79, dar favorizează și efectul patogen al mutațiilor din aceeași genă.78 Un alt exemplu este polimorfismul S1103Y din gena SCN5A, întâlnit mai ales la persoanele de culoare, care are o incidență de aproape 13% și este asociat cu un risc crescut de moarte subită în copilărie.80
În mod interesant, în produsul genei SCN5A, (care codifică izoforma canalului de sodiu Nav1.5 la om) au fost descrise două situsuri alternative de procesare care generează două tipuri de canale de sodiu: unul cu 2016 aminoacizi care conțin glutamină în poziția 1077 (Q1077) și altul cu 2015 aminoacizi lipsiți de glutamină (Q1077del). Transcriptele acestor prelucrări alternative sunt prezente în proporție de 2:1 în aceeași inimă umană și mai multe polimorfisme frecvente vor avea efecte diferite asupra funcționării canalului, în funcție de contextul în care se află Q1077 sau Q1077del. Acest lucru a fost demonstrat inițial cu polimorfismul H558R al SCN5A, prezent la până la 30% din populație. Atunci când H558R a fost exprimat în contextul Q1077, s-a observat o reducere profundă a curentului ionic.81 Un efect similar a fost documentat cu polimorfismul S524Y82. Aceste constatări au furnizat factori care să explice severitatea variabilă a bolii, precum și fenotipurile diferite ale aceleiași mutații observate în unele familii.77
Testarea farmacologică cu adrenalină
Testarea farmacologică cu doze mici de adrenalină este o opțiune sigură și utilă pentru a demasca cazurile suspecte de LQTS cu un QTc la limită. Este deosebit de eficient pentru detectarea formelor asimptomatice de LQTS1, cu o sensibilitate de 92,5%, o specificitate de 86%, o valoare predictivă pozitivă de 76% și o valoare predictivă negativă de 96%. Poate fi, de asemenea, util în diagnosticul LQTS2, cu o sensibilitate și o specificitate mai scăzute. Nu este util pentru LQTS3 sau alte forme de LQTS. În condiții normale, stimularea simpatică induce fosforilarea canalului de potasiu IKs, optimizând funcția acestuia și dând naștere la scurtarea potențialului de acțiune. La pacienții cu SQTL, în special tipul 1, se observă un răspuns paradoxal la administrarea de adrenalină în doze mici (0,025-0,2 µg/kg/min) care prelungește intervalul QT la mai mult de 30 ms83-86.
PROLUNGIREA INTERVALORULUI QT ȘI TORSADE DE PUNCTE INDUSE DE MEDICAMENTE
O mare varietate de medicamente utilizate în diferite specialități medicale pot determina o creștere iatrogenă a intervalului QT. Unele medicamente au fost retrase de pe piață din cauza acestui efect nedorit (de exemplu, astemizol și cisapridă, printre altele; pentru mai multe informații, vizitați www.qtdrugs.org).87,88
Aritmia ventriculară secundară medicamentelor non-antiaritmice apare la mai puțin de unul din 10 000 până la 100 000 de subiecți expuși. Având în vedere că studiile clinice includ între 2 000 și 3 000 de subiecți, acest eveniment advers nedorit și fatal ar scăpa cu ușurință de detectare în timpul fazei clinice de dezvoltare a medicamentului.89 Acest aspect a generat un interes enorm pentru aspectele care se referă la siguranța în studiul și dezvoltarea de noi medicamente.
Factorii legați de susceptibilitatea individuală includ sexul feminin, hipocalcemia, hipomagneziemia, bradicardia, insuficiența cardiacă, postcardioversia, fibrilația atrială, hipertrofia ventriculară stângă, LQTS nedetectată, polimorfismele predispozante și concentrațiile serice ridicate ale medicamentelor predispozante.90
Canalul care interacționează de obicei cu medicamentele este IKr, codificat de gena KCNH2(HERG), datorită structurii sale moleculare. Alte canale de potasiu au 2 resturi de prolină înclinate spre porul canalului, reducând lumenul acestuia. În schimb, IKr este lipsit de aceste reziduuri, se generează un vestibul mai mare al porului și este facilitată expunerea la moleculele mari. În plus, are 2 reziduuri aromatice (tirozină și fenilalanină) care favorizează legarea cu moleculele aromatice prezente în mai multe medicamente capabile să blocheze canalul.91
Cum s-a menționat mai sus, penetranța LQTS este incompletă și unii purtători asimptomatici de mutații ar putea manifesta aritmie malignă la administrarea unuia dintre aceste medicamente. În plus, polimorfismele considerate frecvente în populație conferă susceptibilitate individuală la apariția torsadei de pointes atunci când sunt utilizate unele medicamente. Acesta este cazul polimorfismului R1047L, al doilea cel mai frecvent în KCNH2, care a fost asociat cu dezvoltarea torsadei de pointes la utilizarea medicamentului dofetilidă.92 Cel puțin 20 de polimorfisme ale genei KCNH2 au fost descrise la persoane sănătoase, iar efectul lor în susceptibilitatea individuală de a dezvolta aritmii legate de medicamente rămâne de determinat.93 Polimorfismele care conferă susceptibilitate la dezvoltarea aritmiei ventriculare au fost documentate și în canalul de sodiu Na1.5. Este cazul polimorfismului H558R, care este prezent la până la 30% din populație, sau S1103Y, care este frecvent la persoanele de culoare80,81,90,90,94,95; implicarea lor în susceptibilitatea indusă de medicamente nu a fost investigată.
SINDROMUL QT LUNG ȘI GRĂDINIȚA
Consilierea genetică este importantă în SQTL, dar, în termeni generali, nu există o contraindicație pentru sarcină la femeile purtătoare, deși fiecare caz este diferit și trebuie evaluat individual în contextul adecvat.
A fost observat că riscul de a prezenta aritmie ventriculară malignă scade odată cu sarcina. În schimb, a fost raportată o vulnerabilitate mai mare de a prezenta aritmie malignă în primele 9 luni după naștere, în special la pacientele cu SQTL2. Acest risc scade considerabil cu terapia cu beta-blocante.96
STRATIFICAREA RISCULUI
Evoluția LQTS variază și este influențată de durata intervalului QTc, factorii de mediu, vârsta, genotipul și răspunsul la tratament.97,98 Aritmia ventriculară este mai frecventă în LQTS1 și LQTS2, dar este mai severă în LQTS3.99 După cum a fost menționat mai sus, femeile sunt deosebit de susceptibile la aritmie malignă în perioada postpartum.14
Sindromul QT lung trebuie considerat de risc ridicat atunci când este asociat cu următoarele:
1. Surditate congenitală (sindromul Jervell-Lange-Nielsen).
2. Sincopă recurentă datorată tahiaritmiei ventriculare maligne.
3. Antecedente familiale de moarte subită.
4. QTc>500 ms.
5. Bloc atrioventricular 2:1.
6. Blocaj atrioventricular 2:1.
6. Alternanță electrică a undei T.
7. Genotipul LQTS3.
Studiul lui Priori et al97 realizat pe 647 de pacienți a arătat că probabilitatea de a prezenta un eveniment major (sincopă, stop cardiac, moarte subită) înainte de 40 de ani este mare (>50%) atunci când QTc este >500 ms în LQTS1, LQTS2 și la bărbații cu LQTS3. Recent, a fost raportată o analiză a registrului internațional LQTS. Riscul de moarte subită a fost analizat la 2772 de adolescenți cu această boală și au fost identificați 3 factori asociați cu un risc mai mare la această populație: QTc>530 ms, antecedente de sincopă în ultimii 10 ani și sexul; băieții cu vârste cuprinse între 10 și 12 ani au avut un risc mai mare decât fetele, dar în intervalul de vârstă 13-20 de ani, riscul a fost comparabil.100
TRATAMENT
Pacienții simptomatici care nu primesc tratament au o rată anuală de mortalitate de 20% și o mortalitate la 10 ani de 50% după un prim eveniment de aritmie ventriculară. Deși este clar că tratamentul trebuie instituit atunci când există simptome, abordarea care trebuie utilizată la pacienții asimptomatici este încă în dezbatere. S-a documentat faptul că stopul cardiac poate fi prima manifestare a bolii la 9% dintre pacienți,48 și că 12% dintre pacienții asimptomatici vor dezvolta simptome și pot suferi moarte subită. Tratamentul inițial cu beta-blocante trebuie inițiat la toți pacienții cu SQTL. Restricția la efort este recomandabilă, dar markerii de risc clinic și electrocardiografic reprezintă o bază utilă pentru luarea deciziilor. Este important să se informeze pacienții cu privire la riscul utilizării mai multor medicamente care pot prelungi intervalul QT și favoriza apariția aritmiei ventriculare, așa cum se menționează mai sus. Diagnosticul genetic, pe lângă faptul că permite o consiliere familială adecvată legată de boală, este un ajutor pentru evaluarea prognosticului și orientarea tratamentului specific.
Beta-blocante
Beta-blocantele reprezintă tratamentul de primă linie pentru SQTL și toți pacienții ar trebui să le primească ca terapie inițială.101 Aceștia asigură o reducere a riscului de evenimente cardiovasculare de până la 64%100 și sunt deosebit de eficienți la pacienții cu mutații ale canalelor IKs (LQTS1),102 care sunt reglate în mare măsură de sistemul simpatic. Beta-blocantele nu modifică intervalul QT, ci dispersia acestuia.103 Deși aceste medicamente scad incidența evenimentelor,104,105 s-a demonstrat că 10% dintre pacienții cu LQTS1, 23% cu LQTS2 și 32% cu LQTS3 vor avea simptome cardiovasculare în ciuda tratamentului.106 Pacienții cu LQTS3, în special, nu par să obțină beneficii importante; de fapt, acest grup de medicamente trebuie utilizat cu prudență la acești pacienți, deoarece episoadele de aritmie ventriculară în LQTS3 sunt mai frecvente atunci când frecvența cardiacă este scăzută. În termeni generali, 32% dintre pacienții simptomatici vor avea simptome recurente în primii 5 ani înainte de începerea tratamentului cu beta-blocante, iar 14% dintre pacienții salvați de un episod de moarte subită vor prezenta un alt eveniment similar în decurs de 5 ani dacă vor primi doar acest tratament. 107 În tratamentul SQTL au fost utilizate mai multe beta-blocante, în principal nadolol (0,5-1 mg/kg/zi), propranolol (2-4 mg/kg/zi), metoprolol (0,5-1 mg/kg/zi) și atenolol (0,5-1 mg/kg/zi). Cu toate acestea, atenololul poate să nu fie benefic în SQTL; a fost notificat faptul că cel puțin 75% dintre pacienții care nu au răspuns la tratamentul cu beta-blocante primeau atenolol, deși această constatare poate fi legată de utilizarea unor doze suboptime. 104 Testul de efort este util pentru a stabili doza adecvată. Frecvența cardiacă maximă nu trebuie să depășească 130 bătăi/min în timpul tratamentului.
Blocanți ai canalelor de sodiu
Mutațiile canalului de sodiu care cauzează LQTS3 produc inactivarea defectuoasă a canalului; blocajul canalului de sodiu s-a dovedit a fi util la acești pacienți. Studiile efectuate cu flecainidă au documentat îmbunătățiri ale frecvenței cardiace, ale modificărilor undei T și ale intervalului QT.108 S-a raportat, de asemenea, că mexiletina îmbunătățește markerii electrocardiografici de risc.63,109,110 Studiile in vitro cu ranolazină au arătat scăderi ale efectelor dăunătoare ale mutațiilor raportate la om.111 Deși rezultatele sunt încurajatoare, trebuie avut în vedere faptul că nu există studii pe termen lung care să evalueze această terapie și nici rezultate raportate din serii mari. Blocantele canalelor de sodiu nu trebuie administrate dacă nu există un diagnostic genetic confirmat.
Suplimentele de potasiu și medicamentele care cresc disponibilitatea acestuia
Suplimentele de potasiu și/sau medicamentele care economisesc potasiul, cum ar fi spironolactona, scurtează intervalul QTc în 24% din cazuri.112,113 Medicamentele care favorizează deschiderea canalelor de potasiu, cum ar fi aprikalim, levcromakalim, nicorandil și pinacidil, s-au dovedit a fi utile în tratamentul SQTL. Subtipurile în care acestea sunt deosebit de benefice sunt LQTS1 și LQTS2.114
Stimularea pacemakerilor și defibrilatoarelor
Stimularea pacemakerilor a fost utilizată la pacienții cu aritmie dependentă de pauză.115,116 Pacienții cu LQTS3 beneficiază de obicei mai mult de acest tratament deoarece prevalența bradicardiei este mai mare în acest grup. Stimularea DDD este indicată la pacienții cu aritmie dependentă de pauză sau bloc AV de grad înalt 2:1. Frecvențele programate sub 70 bătăi/min117 nu sunt utile pentru prevenirea aritmiei ventriculare. Se recomandă programarea senzorului la răspuns rapid, deoarece acești pacienți au de obicei o accelerare necorespunzătoare a ritmului cardiac ca răspuns la efort. Toate funcțiile care implică prezența pauzelor trebuie să fie dezactivate, cum ar fi funcția de histerezis și funcția nocturnă. PARP (perioada refractară atrială postventriculară) trebuie să fie cât mai scurtă posibil. Funcția de reglare a frecvenței ar trebui să fie activată pentru a preveni pauza postextrasistolică. Trebuie reamintit faptul că suprasensibilitatea undei T și eșecurile de captare pot, de asemenea, să dea naștere la pauze. Utilizarea combinată a unui defibrilator cardioverter implantabil (ICD) și a beta-blocantelor scade substanțial incidența morții subite.118-120 Indicația acestor măsuri este clară în cazurile cu risc ridicat.121 Programarea dispozitivului va varia în funcție de nevoile fiecărui pacient în parte, dar, în general, trebuie evitată administrarea tratamentului în cazul evenimentelor asimptomatice, autolimitate; în acest scop, este indicat un timp de detecție de 15 s. Furtuna aritmică este o complicație a tratamentului cu AID. Aproape 15% dintre pacienți pot prezenta această complicație, care se datorează, în bună parte, tonusului simpatic crescut în urma șocului ICD.118 Această problemă poate fi gestionată prin creșterea dozei de beta-blocante. Dacă această măsură nu este utilă, trebuie luată în considerare rezecția ganglionilor lanțului simpatic.
Simpaticectomia stângă
În 1971, ganglionectomia simpatică a fost introdusă ca o opțiune terapeutică utilă la acești pacienți.122 În 1991, Schwartz et al123 au publicat prima serie de 85 de pacienți cu un răspuns slab la tratamentul cu beta-blocante, la care s-a efectuat o stellectomie stângă cu rezultate încurajatoare: o rată de supraviețuire la 5 ani de 94%. În prezent, această opțiune terapeutică este oferită pacienților cu risc ridicat care persistă cu sincope în ciuda tratamentului cu beta-blocante și/sau a implantării stimulatorului cardiac, precum și celor care prezintă șocuri frecvente de la defibrilatorul implantat. Procedura constă în rezecția porțiunii inferioare a ganglionului stelat și a ganglionilor toracici stângi T2 până la T4 din lanțul simpatic, deoarece stellectomia stângă simplă nu s-a dovedit a fi suficient de eficientă. Toracoscopia microinvazivă124,125 a fost utilizată cu rezultate bune. Cea mai mare serie de pacienți tratați prin această metodă a fost raportată recent și a arătat o reducere semnificativă a numărului de episoade de sincopă sau de moarte subită, precum și o rată de supraviețuire la 5 ani de 95%. La pacienții cu sincope anterioare, supraviețuirea la 5 ani a fost de 97%, cu o posibilitate de recidivă de 11%, care, în majoritatea cazurilor, a constat într-un singur eveniment sincopal. A existat, de asemenea, o reducere semnificativă a segmentului QT în urma simpatectomiei stângi. În ciuda acestor rezultate favorabile, prevenirea morții subite nu este completă, dar a fost redusă la 3%. La pacienții cu un ICD care au fost supuși unei intervenții chirurgicale din cauza șocurilor multiple ale defibrilatorului, numărul mediu de evenimente a scăzut de la 25 la 0, o reducere de 95%. Un efect benefic a fost confirmat în LQTS1. Este probabil ca beneficiile să fie mai mici la pacienții cu LQTS2, iar la LQTS3, eficacitatea sa nu a fost dovedită.126
Ablația
A fost raportat faptul că ablația extrasistolei, care în unele cazuri inițiază aritmia ventriculară, poate fi efectuată cu o reducere a incidenței episoadelor.127 Cu toate acestea, nu există studii pe termen lung cu un număr adecvat de pacienți care să justifice utilizarea de rutină a acestei tehnici.
Vezi editorialul de la paginile 675-82
ABREVIERI
AV: atrioventriculară
AID: defibrilator automat implantabil
ECG: electrocardiogramă
QTc: QT corectat în funcție de frecvența cardiacă
ATS: Sindromul Andersen-Tawil
LQTS: sindromul QT lung
Dr. Medeiros beneficiază de sprijin economic din partea CONACyT și FUNSALUD.