AML:n ja CML:n luokittelu
AML:n ranskalais-amerikkalais-brittiläinen (FAB) luokitus perustui sytomorfologisiin piirteisiin, ja se on korvattu WHO:n luokituksilla 2001/2008 ja 2016.
Euroopan LeukemiaNet (ELN) määrittelee 3 riskiryhmää geneettisten poikkeavuuksien mukaan (Döhner ym. 2017).
Tietyt AML:n alaryhmät, kuten akuutti promyelosyyttinen leukemia (APL) (PML-RARA, t) hyötyvät kohdennetusta hoidosta ja niiden ennuste on erinomainen.
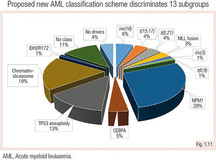
Ehdotettiin uutta luokituskaaviota, joka sisältää karyotyypin ja somaattiset mutaatiot, ja siinä määritellään 13 AML:n alaryhmää (Papaemmanuil ym. 2016).
Spesifiset kromosomipoikkeavuudet, kuten t(8;21), inv(16), t(15;17), ovat tautia määritteleviä riippumatta kvantifioidusta blastimäärästä.
Tulevaisuudessa AML:n diagnoosi saattaa nojautua pelkästään geneettisiin löydöksiin.
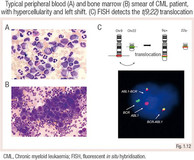
CML:lle on ominaista perifeerisessä veressä esiintyvä perifeerisen veren myelooisten progenitoreiden sisältämä, ”vasemmanpuoleiseksi siirtymäksi” kutsuttu leukosytoosi. Taudin taustalla on Philadelphia-kromosomi t(9;22), joka tuottaa konstitutiivisesti aktiivista fuusioproteiinia BCR-ABL1.
CML luokitellaan krooniseen vaiheeseen, kiihtyvään vaiheeseen ja blastivaiheeseen blastisolujen lukumäärän mukaan.
Terapian seurannassa käytetään erittäin herkkää reaaliaikaista PCR:ää, jolla osoitetaan BCR-ABL1.
Tarkistuskysymykset
- Mihin WHO:n vuoden 2016 AML-luokitus perustuu?
- Mitkä aberraatiot ja mutaatiot ovat AML:n diagnostiikkaa ilman, että tarvitaan ≥20 %:n blastisoluja?
- Mitkä ovat CML:n geneettiset perusteet?