INTRODUZIONE
La sindrome del QT lungo (LQTS) è caratterizzata da una ripolarizzazione ventricolare gravemente alterata, con conseguente prolungamento dell’intervallo QT sull’elettrocardiogramma (ECG). La condizione predispone i pazienti all’aritmia ventricolare maligna (torsade de pointes) e alla morte improvvisa. La descrizione clinica ed elettrocardiografica della sindrome del QT lungo fu riportata nel 1957 da Anton Jervell e Fred Lange Nielsen,1 che pubblicarono i loro studi su una famiglia di genitori non consanguinei con 6 figli. Quattro dei bambini avevano sordità congenita ed episodi sincopali, e 3 presentavano morte improvvisa. Lo studio ECG di questi pazienti ha mostrato un intervallo QT insolitamente lungo. Entrambi i genitori erano asintomatici, avevano un ECG normale e non presentavano problemi di udito. Nel 1964, Romano e Ward hanno riportato indipendentemente una sindrome cardiaca caratterizzata da sincope ricorrente, una storia familiare di morte improvvisa e un prolungamento dell’intervallo QT senza sordità neuronale.2 Studi genetici successivi hanno dimostrato che la sindrome descritta da Jervell e Lange Nielsen, che è accompagnata da sordità neuronale congenita, corrisponde a mutazioni omozigoti, con un fenotipo grave e alto rischio di morte improvvisa. La condizione nota come sindrome di Romano-Ward corrisponde generalmente a mutazioni eterozigoti, i pazienti non mostrano alterazioni dell’udito e la gravità della malattia varia notevolmente. Quasi mezzo secolo dopo, nel 1995,3,4 sono stati descritti i principali geni associati alla LQTS e la malattia è stata riconosciuta come un disturbo dei canali ionici cardiaci. È stata la prima canalopatia cardiaca ad essere descritta ed è forse il disturbo del canale ionico aritmogeno più ampiamente studiato fino ad oggi. Il quadro clinico varia notevolmente: il paziente può essere asintomatico o mostrare sincopi ricorrenti, convulsioni o morte improvvisa come prima manifestazione della malattia. Inizialmente, la LQTS era considerata una sindrome rara e, in effetti, la presentazione grave della malattia è sporadica. Tuttavia, l’incidenza delle mutazioni correlate è stimata in 1/3000-5000 casi,5 il 32% dei portatori asintomatici può avere un intervallo QT corretto per la frequenza cardiaca (QTc) entro i limiti normali, la malattia viene trasmessa al 50% dei loro discendenti, sono più suscettibili a sviluppare aritmie rispetto alla popolazione generale, e fino al 20% può diventare sintomatico.6
La sindrome del QT lungo mostra una grande eterogeneità genetica. Più di 500 mutazioni distribuite in 10 geni sono state descritte in questa condizione: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 e SCN4B. Nonostante i progressi in questo settore, una diagnosi genetica non può essere stabilita nel 25%-30% dei pazienti.7,8 La presentazione della malattia è principalmente monogenica6; le varietà poligeniche o composite hanno solitamente un fenotipo più grave. La penetranza, cioè i pazienti che hanno la mutazione e manifestano il fenotipo, varia dal 25% al 90%.9 Meno frequentemente, ci possono essere variazioni nell’espressività della malattia, con diversi fenotipi derivanti dalla stessa mutazione. Gli studi di genetica molecolare sviluppati negli ultimi 11 anni hanno prodotto importanti correlazioni genotipo-fenotipo, che hanno contribuito a guidare l’approccio terapeutico. Inoltre, interessanti osservazioni sono state fatte sulla suscettibilità individuale a sviluppare l’aritmia negli studi che indagano i frequenti polimorfismi non sinonimi in questa popolazione, un aspetto che ha suscitato notevole interesse, in particolare nell’area della farmacogenomica.
CLASSIFICAZIONE DELLA SINDROME DEL QT LUNGO
Concetti generali
La classificazione della LQTS utilizzata in passato si basava sulla presentazione omozigote o eterozigote della malattia, che dà origine rispettivamente alla sindrome di Jervell-Lange-Nielsen (con sordità) e alla sindrome di Romano-Ward (senza sordità). La presente classificazione enfatizza i risultati genetici, come illustrato nella Tabella 1. I 3 geni principali associati alla malattia sono stati descritti nel 1995-1996. Questi geni, che codificano per le unità formanti pori dei canali del potassio IKs e IKr, e il canale del sodio Nav1.5, rappresentano quasi il 65% dei casi. Anche se negli anni successivi altri sette geni sono stati inclusi nella lista, essi rappresentano solo il 5% dei casi.
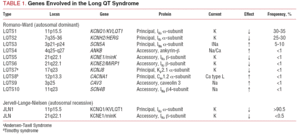
I canali ionici sono proteine transmembrana che trasportano ioni attraverso la membrana cellulare. I canali implicati nella LQTS sono selettivi o specializzati nel trasporto di un singolo ione e sono voltaggio-dipendenti, cioè la loro attivazione avviene ad una specifica tensione intracellulare, che varia a seconda del sottotipo di canale. I fenomeni elettrici e contrattili che si verificano nel cardiomiocita sono controllati da queste strutture. I canali ionici formano complessi macromolecolari composti da un’unità principale che forma il poro del canale e proteine ausiliarie che lo regolano (Figura 1). La disfunzione del canale vista nella LQTS può verificarsi in questi due siti: la proteina principale o le proteine regolatrici (Tabella 1). Il coinvolgimento dell’unità che forma i pori, nota come alfa, genera i tre sottotipi più comuni di LQTS: LQTS1 (che colpisce il canale del potassio IKs), LQTS2 (che colpisce il canale del potassio IKr), e LQTS3 (che colpisce il canale del sodio). Poiché questi sono i sottotipi più frequenti, sono i meglio caratterizzati clinicamente e geneticamente. Le correlazioni fenotipo-genotipo in queste tre forme principali sono descritte nella Figura 2. Attualmente, la sindrome di Jervell-Lange-Nielsen corrisponde alle varietà LQTS 1 e 5. Caratteristicamente, questi pazienti hanno sordità congenita e mutazioni composte omozigoti o eterozigoti che influenzano la corrente IKs. La sindrome di Romano-Ward comprende le varietà da LQTS 1 a 10 e non comporta sordità.
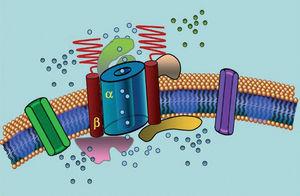
Figura 1. Rappresentazione schematica del complesso macromolecolare. I canali ionici sono proteine transmembrana (α) regolate da varie proteine; una di queste è la cosiddetta subunità β.
Sindrome del QT lungo di tipo 1 (LQTS1)
I pazienti con LQTS1 di solito presentano episodi di aritmia ventricolare durante l’esercizio fisico o quando sono sottoposti a stimolo simpatico (68%).10 Il nuoto è stato descritto come uno sport che scatena l’aritmia nella LQTS1.11 La penetrazione è quasi del 62% in questo sottotipo. L’onda T in questi pazienti ha spesso una base ampia e una durata molto prolungata12,13 (Figura 2). È il sottotipo più frequente e spiega il 30%-35% dei casi. Il gene interessato, KvLQT1 (o KCNQ1), si trova sul cromosoma 11 (11p15.5) e codifica per la subunità α del canale del potassio IKs. Il potenziale d’azione è prolungato da una riduzione della corrente K+ in uscita durante la fase 3 del potenziale d’azione.
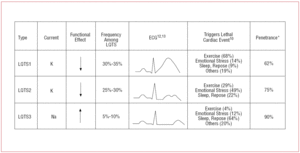
Figura 2. Correlazione genotipo-fenotipo nelle sindromi del QT lungo più frequenti. *Riferimento ai casi che hanno la mutazione e manifestano il fenotipo.
Sindrome del QT lungo di tipo 2 (LQTS2)
I pazienti con LQTS2 tendono a presentare aritmia ventricolare in risposta a stress emotivo (49%) o stimoli uditivi improvvisi (per esempio, una sveglia), e meno frequentemente durante il sonno (22%) o l’esercizio fisico (29%).10 Le donne nel periodo post-partum sono particolarmente suscettibili.14 La penetranza stimata è del 79%; quindi, fino al 20% dei casi può avere un ECG non diagnostico. L’onda T nella LQTS2 è solitamente di bassa ampiezza e bifida, con notching12,13 (Figura 2). Il gene interessato è KCNH2 o HERG, situato sul cromosoma 7 (7q35-36), che codifica per il canale del potassio IKr α-subunità e rappresenta il 25%-30% dei casi. La disfunzione di questo canale diminuisce la corrente K+ in uscita durante la fase 3 del potenziale d’azione, prolungandone la durata.
Sindrome del QT lungo di tipo 3 (LQTS3)
I pazienti con LQTS3 hanno un rischio maggiore di presentare aritmie maligne durante il riposo (sonno) o bradicardia.15 La penetrazione della mutazione del gene SCN5A è quasi del 90%. L’ECG nella LQTS3 di solito mostra un’onda T ritardata e appuntita e permette di osservare chiaramente il prolungamento del segmento ST12,13 (Figura 2). Questi pazienti di solito hanno meno sintomi di quelli con LQTS1 o LQTS2, ma gli eventi sono caratteristicamente più letali.
Il gene interessato nella LQTS3 è SCN5A, che codifica per la subunità α del canale del sodio Nav1.5 (Figura 1), situato sul cromosoma 3 (3p21-24); è la causa della malattia nel 5%-10% dei casi. L’inattivazione difettosa del canale permette un ingresso sostenuto di Na+ durante la fase 2 del potenziale d’azione, prolungandone la durata.
La sindrome del QT lungo di tipo 4 (LQTS4)
Il tipo 4 è una varietà rara di LQTS, che rappresenta quasi l’1% dei casi. Si tratta di una forma atipica che produce un ampio spettro di aritmie, tra cui tachicardia ventricolare polimorfica catecolaminergica, fibrillazione atriale, alterazioni della conduzione intraventricolare, disfunzione del nodo del seno e bradicardia6-18; inoltre, il QTc può essere entro limiti normali in molti pazienti. Il gene interessato è ANKB, situato sul cromosoma 4 (4q25-27), che codifica per la sintesi di ankyrin-β, una proteina strutturale che collega le proteine di membrana dei cardiomiociti alle proteine del citoscheletro. Queste proteine sono la pompa Na/K ATPasi, lo scambiatore Na/Ca e il recettore dell’inositolo trifosfato (InsP3R). Le mutazioni che causano una perdita della funzione ankyrin-β portano ad aumenti della concentrazione intracellulare di calcio e ad alterazioni nell’espressione della N/K ATPasi e dello scambiatore Na/Ca. L’elevata concentrazione di calcio dà luogo a post-depolarizzazioni precoci e ritardate. Così, le aritmie ventricolari osservate nelle mutazioni del gene ankyrin-β sono dovute a depolarizzazioni spontanee, di solito in risposta alla stimolazione catecolaminergica.
La sindrome del QT lungo di tipo 5 (LQTS5)
Il tipo 5 ha origine da cambiamenti nella sequenza del gene KCNE1 situato sul cromosoma 21 (21q22.1p22.)19 KCNE1 codifica per la sintesi della subunità β del canale IKs, nota anche come subunità minK, che regola il canale IKs. Questo tipo rappresenta meno dell’1% dei casi.
Sindrome del QT lungo di tipo 6 (LQTS6)
Il gene interessato nel tipo 6 è KCNE2, situato sul cromosoma 21 (21q22.1).20 Questo gene codifica per la subunità β del canale del potassio, nota anche come subunità MiRP1, e regola il canale IKr. Meno dell’1% dei casi sono di tipo 6.
Sindrome del QT lungo di tipo 7 o sindrome di Andersen-Tawil (LQTS7)
I reperti dismorfici e le alterazioni elettrocardiografiche di questa sindrome sono stati descritti per la prima volta nel 1971 dal dottor Andersen21 e rivisitati nel 1994 dal dottor Tawil,22 ma la descrizione genetica/molecolare è stata riportata solo nel 2001.23 Ora nota come sindrome di Andersen-Tawil (ATS), questa condizione è un’alterazione autosomica dominante caratterizzata da paralisi periodica, sviluppo scheletrico anomalo, aritmia ventricolare del tipo che comporta frequenti extrasistoli ventricolari e una particolare suscettibilità a sviluppare fibrillazione ventricolare, in particolare nelle donne. Le alterazioni descritte nell’ATS includono extrasistoli ventricolari (41%), tachicardia ventricolare polimorfa non sostenuta (23%), tachicardia ventricolare bidirezionale (68%) e torsade de pointes (3%).24 Alcune delle caratteristiche dismorfiche osservate includono bassa statura, scoliosi, clinodattilia, ipertelorismo, basso impianto delle orecchie, micrognazia e fronte ampia. L’espressione della malattia varia, un fatto che complica la diagnosi precoce.23,25 Le mutazioni nel gene KCNJ2 situato nel cromosoma 17 (17q23), che codifica per la sintesi del canale del potassio rettificante Kir 2.1, rappresentano il 70% dei casi. Questo canale partecipa alla fase 4 del potenziale d’azione. Diversi autori mettono in dubbio l’inclusione di questo gene nel gruppo causale della LQTS, perché l’intervallo QTc è solo leggermente prolungato in questa sindrome o addirittura normale, ma l’onda U è di solito prominente, il che ha portato alla sopravvalutazione dell’intervallo QT. Il lettore troverà che alcuni autori suggeriscono che le mutazioni KCNJ2 generano la ATS1 e non la LQTS7.24
La sindrome del QT lungo di tipo 8 (LQTS8)
Il tipo 8 deriva da mutazioni nel gene CACNA1 situato sul cromosoma 12 (12p13.3), che codifica per il canale del calcio L-tipo Cav1.2. Causa la sindrome di Timothy,26 una condizione caratterizzata da malformazioni cardiache, deficit immunologico intermittente, ipoglicemia, alterazioni cognitive tra cui l’autismo, fusione interdigitale e QT prolungato, che porta ad aritmia cardiaca e morte improvvisa.27 Meno dello 0,5% dei casi sono di tipo 8.
Sindrome del QT lungo di tipo 9 (LQTS9)
Questa varietà di LQTS si sviluppa da mutazioni nel gene CAV3, situato sul cromosoma 3 (3p25), che codifica per la sintesi della caveolina 3. La caveolina è un’invaginazione della membrana plasmatica implicata nell’endocitosi, nell’omeostasi dei lipidi e nella trasduzione del segnale. Un componente importante di questa struttura è la caveolina, che ha 3 sottotipi conosciuti; il sottotipo 3 è specifico per il muscolo scheletrico e cardiaco. Alcuni canali ionici sono co-ubicati nella caveola, compresa un’isoforma cardiaca del canale del sodio Nav1.5. Diverse mutazioni in questa proteina sono state recentemente descritte. Queste alterano le proprietà biofisiche del canale del sodio Nav1.5 in vitro, generando un fenotipo simile a quello osservato nella LQTS3.28 Meno dell’1% dei casi sono attribuiti a questa causa.
La sindrome del QT lungo tipo 10 (LQTS10)
Il tipo 10 è stato descritto in un caso molto grave, con QTc >600 ms, bradicardia fetale e blocco atrioventricolare (AV) 2:1. Risulta da mutazioni nel gene SCN4B, situato sul cromosoma 11 (11q23), che codifica per il canale del sodio β4-subunità. Sono stati descritti quattro diversi sottotipi di subunità β, che interagiscono e regolano le varie isoforme del canale del sodio; tuttavia, solo il sottotipo 4 è stato finora associato all’aritmogenesi.29 L’incidenza delle mutazioni di questo gene non è stata esaminata, ma è stimata a
Mutazioni della varietà Jervell-Lange-Nielsen
Questa forma grave di LQTS è causata da mutazioni omozigoti30 o eterozigoti composti dei geni KCNQ1, e/o KCNE1, che codificano per la corrente IKs; cioè, una varietà molto grave delle forme LQTS1 o LQTS5. Questa condizione è caratteristicamente associata a sordità congenita. I pazienti di solito hanno un QTc>500 ms e sincope ricorrente, e sono ad alto rischio di morte improvvisa. I genitori dei pazienti con questa varietà sono di solito eterozigoti e hanno una malattia meno grave, o non mostrano alcun sintomo.31
DIAGNOSI DELLA SINDROME DEL QT LUNGO
Schwartz Score
Nel 1985, Schwartz et al32 pubblicarono i criteri per la diagnosi della LQTS, che furono modificati nel 1993 e contengono importanti linee guida per la valutazione iniziale dei casi potenziali. Questo sistema utilizza un punteggio da 1 a 9 basato sulla storia familiare e sui risultati clinici ed elettrocardiografici. La probabilità di malattia è bassa con un punteggio ≥1, intermedia con 2-3 e alta con ≥4 (Tabella 2).

Diagnosi prenatale della sindrome del QT lungo
La bradicardia fetale può essere una delle prime manifestazioni cliniche della LQTS. Serie retrospettive hanno dimostrato che fino al 70% dei pazienti con diagnosi di LQTS durante l’infanzia hanno una storia di bradicardia, di solito accompagnata da idrope fetale.33 La valutazione della ripolarizzazione cardiaca fetale tra le settimane 14 e 39 è utile per la diagnosi precoce di LQTS.34
Il mosaicismo gonadico per LQTS è stato associato a perdite fetali ricorrenti durante il terzo trimestre di gravidanza.35 Se la malattia è altamente sospetta, l’amniocentesi dopo 16 settimane di gestazione può essere utile per stabilire la diagnosi, che è facilmente raggiungibile quando uno dei genitori è noto per essere portatore di una mutazione specifica.36
STUDIO DI UN PAZIENTE CON SINDROME DEL QT LUNGO
Anamnesi clinica
Una storia familiare e/o personale di morte improvvisa è di fondamentale importanza sia per la diagnosi che per la stratificazione del rischio della LQTS. Inoltre, i fattori precipitanti e il contesto della sincope possono indicare il sottotipo di LQTS. Nella valutazione iniziale di un caso sospetto, l’uso di farmaci che possono prolungare l’intervallo QT dovrebbe essere escluso.
Intervallo QT: Cosa è normale?
L’intervallo QT deve essere misurato preferibilmente nelle derivazioni II o V5,37 dove è stato dimostrato che ha un maggiore valore predittivo.38 Questo intervallo indica la durata della ripolarizzazione ventricolare e viene misurato dall’inizio dell’onda Q alla fine dell’onda T. Convenzionalmente, si utilizza la formula proposta da Bazett39 per correggere la durata dell’intervallo in base alla frequenza cardiaca (QTc=QT/√RR, espresso in secondi). Anche se la misurazione dell’intervallo QT sembra semplice, in uno studio multicentrico condotto da Viskin et al,40 meno del 40% dei medici diversi dai cardiologi, meno del 50% dei cardiologi e più dell’80% degli specialisti in aritmia sapevano come misurarlo correttamente. È consigliabile che i medici effettuino la misurazione manuale e non si fidino delle misurazioni automatiche, che possono essere utili per altri intervalli, ma sono imprecise nel calcolo dell’intervallo QT. Il QT è un intervallo dinamico e i limiti normali dipendono da diversi fattori. Anche se un intervallo QTc di é440 ms nei maschi e di é460 ms nelle femmine è considerato anormale, si possono trovare sia portatori di mutazioni che individui sani entro questo intervallo (Figura 3). In famiglie con LQTS1, Vincent et al41 hanno dimostrato che nessuno dei casi con un genotipo positivo aveva un QTc470 ms. Monnig et al38 hanno recentemente dimostrato che il QTc>440 ms è sufficiente per individuare i pazienti con mutazioni associate alla LQTS, il QTc>470 ms è utile per identificare i pazienti a rischio di sviluppare sintomi, e il QTc>500 ms si trova nei pazienti sintomatici in trattamento.
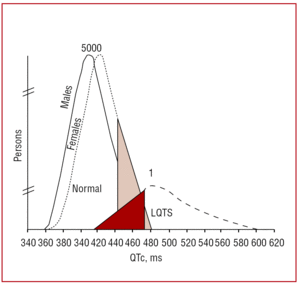
Figura 3. Modello che mostra la distribuzione dell’intervallo QT corretto per la frequenza cardiaca (QTc) in pazienti con mutazioni in KVLQT1, HERG, o SCN5A, e i loro familiari non affetti. La curva a sinistra descrive la distribuzione dei membri non affetti e la curva a destra, i membri affetti.
Altre alterazioni elettrocardiografiche associate alla sindrome del QT lungo
I pazienti con LQTS possono presentare alterazioni multiple dell’onda T: alternanze di polarità, variazioni di ampiezza, notching e un aspetto bifasico, tra gli altri.42 L’alternanza dell’onda T (Figura 4A) è definita come una variazione battito per battito di ampiezza, morfologia e polarità di un’onda T del ritmo sinusale, senza variazioni nel complesso QRS. È un indicatore di instabilità elettrica,43 che riflette la dispersione regionale della ripolarizzazione ventricolare, e occasionalmente precede la fibrillazione ventricolare.44
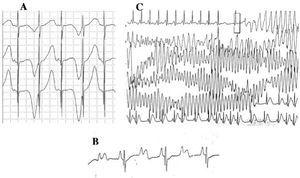
Figura 4. Alterazioni elettrocardiografiche nella sindrome del QT lungo. A: alternanze elettriche dell’onda T. B: blocco atrioventricolare 2:1. C: torsade de pointes autolimitata.
I pazienti con LQTS possono progredire con segni di disfunzione del nodo del seno, bradicardia e/o pause.45 I sottotipi LQTS1 e LQTS3, in particolare quest’ultimo, presentano spesso bradicardia sinusale,46 mentre LQTS4 è stato associato a disfunzione del nodo del seno.18
Dal decennio 1970-1980, è stata osservata la coesistenza di difetti di conduzione AV con LQTS47 (Figura 4B). Il blocco AV due a uno è una manifestazione infrequente con una prognosi sfavorevole che può essere presente fin dallo stadio fetale sotto forma di bradicardia persistente. L’incidenza di questa anomalia è stata riportata al 4%-5%48 ed è associata a un’elevata mortalità nonostante il trattamento con beta-bloccanti e/o pacemaker.49,50 Questo fenomeno può essere spiegato da una lunga durata del potenziale d’azione. Quando il periodo refrattario ventricolare è prolungato, l’impulso seguente dell’attività sinusale è bloccato perché raggiunge i ventricoli quando sono ancora nel periodo refrattario. Questa alterazione sembra verificarsi esclusivamente nella LQTS, perché il periodo refrattario ventricolare è maggiore di quello del sistema di conduzione AV.51 La pendenza del complesso QRS è solitamente ripida e il blocco è stato localizzato nell’area infraHis,46,51,52 ma la sede del blocco può dipendere dal genotipo. Finora, 4 geni sono stati correlati al blocco 2:1 nella LQTS: HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 e SCN4B (LQTS10).55
La caratteristica aritmia ventricolare della LQTS è conosciuta come torsade de pointes (Figura 4C). Si presenta quando l’intervallo QT è prolungato, indipendentemente dall’eziologia. Si tratta di una tachicardia ventricolare polimorfica dovuta al rientro, caratterizzata elettrocardiograficamente da una continua torsione dell’asse QRS intorno a una linea immaginaria. È comunemente preceduta da una pausa seguita da un’extrasistole (intervallo RR breve-lungo-corto), come mostrato nella figura.56-58 Può culminare in fibrillazione ventricolare e morte improvvisa. Se questo non si verifica, il paziente può solo sperimentare una sincope, e se l’episodio è breve, può passare inosservato.
Lo studio Holter
Holter fornisce una valutazione completa e dinamica dell’intervallo QT. Occasionalmente vengono registrati episodi spontanei di aritmia ventricolare asintomatica, così come episodi di disfunzione del nodo del seno o di blocco AV.
Test da sforzo
I pazienti con LQTS non possono raggiungere la frequenza cardiaca massima prevista calcolata in base all’età. Inoltre, sotto sforzo l’intervallo QT può mostrare un comportamento paradossale, aumentando invece di diminuire.59,60 Il pattern elettrocardiografico durante il test da sforzo sarà diverso a seconda del tipo di LQTS. I pazienti con LQTS1, oltre a non raggiungere la frequenza cardiaca massima calcolata per la loro età, mostrano spesso un aumento dell’intervallo QT, mentre quelli con LQTS2 possono raggiungere la frequenza cardiaca prevista e mostrare solo un lieve aumento dell’intervallo QT o nessuno.61,62 In generale, i pazienti con LQTS3 hanno una risposta fisiologica all’esercizio, cioè un normale accorciamento dell’intervallo QT.63 Il test da sforzo può anche essere utile per valutare la risposta al trattamento e per stratificare il rischio nei casi asintomatici, o quando ci sono dubbi sugli eventi che portano all’aritmia.
Screening genetico
Negli ultimi anni, gli studi genetici nella LQTS sono stati limitati ai laboratori di ricerca. Tuttavia, le informazioni derivate da questi sforzi sono state estremamente utili per il trattamento dei pazienti, in particolare i casi ad alto rischio. Forse la principale applicazione dello screening è nella consulenza genetica, ma ha anche importanti implicazioni nel trattamento, che può essere orientato in base al canale interessato. La localizzazione precisa di una data mutazione può fornire ulteriori informazioni sull’evoluzione del rischio. I pazienti con mutazioni nella regione transmembrana di KCNQ1 (IKS) hanno una maggiore probabilità di presentare eventi aritmici rispetto a quelli con mutazioni nella regione C-terminale64 ; lo stesso vale per i pazienti con mutazioni nella regione del poro di KCNH2 o HERG65 rispetto a quelli con mutazioni nelle regioni N o C-terminali.66
Lo screening iniziale può forse essere limitato ai geni KCNQ1, HERG e SCN5A, che offrono la possibilità di incontrare mutazioni nel 65% dei casi. Quando i risultati ottenuti sono negativi, lo screening può essere esteso ai geni KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 e SCN4B, che aumenteranno la possibilità di risultati positivi dal 5% al 10%.
Screening genetico post mortem
È interessante che mutazioni genetiche che portano alla LQTS sono state trovate in bambini che hanno avuto una morte improvvisa e in casi inspiegabili di morte improvvisa in giovani adulti.
Studi genetici post mortem di pazienti con morte improvvisa e autopsia negativa hanno mostrato mutazioni che portano alla LQTS in percentuali variabili67-69: vicino al 10% nei bambini e il 35% nei giovani adulti.70-72 Sulla base di questi risultati, è stato proposto lo studio di routine dell’ECG in tutti i neonati.73,74
Lo studio genetico post mortem, noto anche in letteratura come “autopsia molecolare”, oltre alle ripercussioni legali, ha importanti implicazioni nelle famiglie che potrebbero essere colpite senza saperlo.
Polimorfismi regolatori
Nella popolazione LQTS sono stati descritti diversi polimorfismi frequenti, distribuiti in quasi tutti i geni associati a questa condizione. Anche se questi cambiamenti non sono apparentemente patogeni, alcuni possono avere i seguenti effetti75-78:
1. Generare la suscettibilità individuale a sviluppare l’aritmia.
2. 2. Favorire l’impatto patogeno di un altro cambiamento non sinonimo.
3. Diminuire l’effetto patogeno di un altro cambiamento non sinonimo.
Questo è il caso del polimorfismo K897T in KCNH2 (HERG), che è presente fino al 15% della popolazione e non solo è legato alla suscettibilità a certi farmaci,79 ma favorisce anche l’effetto patogeno delle mutazioni nello stesso gene.78 Un altro esempio è il polimorfismo S1103Y nel gene SCN5A, presente soprattutto nei neri, che ha un’incidenza di quasi il 13% ed è associato ad un aumentato rischio di morte improvvisa nell’infanzia.80
È interessante notare che due siti di elaborazione alternativi che generano due tipi di canali del sodio sono stati descritti nel prodotto del gene SCN5A, (che codifica per l’isoforma del canale del sodio Nav1.5 nell’uomo): uno con 2016 aminoacidi contenenti glutamina nella posizione 1077 (Q1077), e un altro con 2015 aminoacidi senza glutamina (Q1077del). I trascritti di queste elaborazioni alternative sono presenti in una proporzione 2:1 nello stesso cuore umano e diversi polimorfismi frequenti avranno effetti diversi sul funzionamento del canale, a seconda che il contesto sia Q1077 o Q1077del. Questo è stato inizialmente dimostrato con il polimorfismo H558R di SCN5A, presente fino al 30% della popolazione. Quando H558R è stato espresso nel contesto di Q1077, è stata osservata una profonda riduzione della corrente ionica.81 Un effetto simile è stato documentato con il polimorfismo S524Y82. Questi risultati hanno fornito fattori per spiegare la diversa gravità della malattia, così come i diversi fenotipi della stessa mutazione osservati in alcune famiglie.77
Test farmacologico con adrenalina
Il test farmacologico con adrenalina a basse dosi è un’opzione sicura e utile per smascherare casi sospetti di LQTS con un QTc borderline. È particolarmente efficace per rilevare forme asintomatiche di LQTS1, con una sensibilità del 92,5%, specificità dell’86%, valore predittivo positivo del 76% e valore predittivo negativo del 96%. Può anche essere utile nella diagnosi di LQTS2, con sensibilità e specificità inferiori. Non è utile per LQTS3 o altre forme di LQTS. In condizioni normali, la stimolazione simpatica induce la fosforilazione del canale del potassio IKs, ottimizzando la sua funzione e dando luogo all’accorciamento del potenziale d’azione. Nei pazienti con LQTS, in particolare il tipo 1, si osserva una risposta paradossale alla somministrazione di adrenalina a basso dosaggio (0,025-0,2 µg/kg/min) che prolunga l’intervallo QT a più di 30 ms83-86.
PROLUNGAMENTO DELL’INTERVALLO QT E TORSADE DE POINTES INDOTTO DA FARMACI
Una grande varietà di farmaci usati in diverse specialità mediche può causare un aumento iatrogeno dell’intervallo QT. Alcuni farmaci sono stati rimossi dal mercato a causa di questo effetto indesiderato (ad esempio, astemizolo e cisapride, tra gli altri; per ulteriori informazioni, visitare www.qtdrugs.org).87,88
L’aritmia ventricolare secondaria a farmaci non antiaritmici si verifica in meno di uno ogni 10 000-100 000 soggetti esposti. Considerando che gli studi clinici includono tra 2000 e 3000 soggetti, questo evento avverso indesiderato e fatale sfuggirebbe facilmente alla rilevazione durante la fase clinica di sviluppo del farmaco.89 Questo punto ha generato un enorme interesse per gli aspetti relativi alla sicurezza nello studio e nello sviluppo di nuovi farmaci.
I fattori relativi alla suscettibilità individuale includono il sesso femminile, l’ipocalcemia, l’ipomagnesiemia, la bradicardia, l’insufficienza cardiaca, la postcardioversione, la fibrillazione atriale, l’ipertrofia ventricolare sinistra, la LQTS non rilevata, i polimorfismi predisponenti e alte concentrazioni nel siero di farmaci predisponenti.90
Il canale che tipicamente interagisce con i farmaci è IKr, codificato dal gene KCNH2(HERG), a causa della sua struttura molecolare. Altri canali del potassio hanno 2 residui di prolina angolati verso il poro del canale, riducendo il suo lume. Al contrario, IKr manca di questi residui, si genera un vestibolo del poro più grande e si facilita l’esposizione a grandi molecole. Inoltre, ha 2 residui aromatici (tirosina e fenilalanina) che favoriscono il legame con le molecole aromatiche presenti in diversi farmaci in grado di bloccare il canale.91
Come è stato menzionato sopra, la penetranza della LQTS è incompleta e alcuni portatori asintomatici di mutazioni potrebbero manifestare aritmia maligna dopo aver ricevuto uno di questi farmaci. Inoltre, polimorfismi considerati frequenti nella popolazione conferiscono suscettibilità individuale allo sviluppo di torsade de pointes quando alcuni farmaci vengono utilizzati. Questo è il caso del polimorfismo R1047L, il secondo più frequente in KCNH2, che è stato associato allo sviluppo di torsade de pointes con l’uso del farmaco dofetilide.92 Almeno 20 polimorfismi del gene KCNH2 sono stati descritti in persone sane e il loro effetto nella suscettibilità individuale a sviluppare aritmie correlate ai farmaci deve ancora essere determinato.93 Polimorfismi che conferiscono suscettibilità allo sviluppo di aritmia ventricolare sono stati documentati anche nel canale del sodio Na1.5. È il caso del polimorfismo H558R, presente fino al 30% della popolazione, o S1103Y, frequente nei neri80,81,90,94,95; La loro implicazione nella suscettibilità indotta da farmaci non è stata studiata.
SINDROME DEL QT LUNGO E PREGARAZIONE
La consulenza genetica è importante nella LQTS, ma in termini generali non vi è alcuna controindicazione alla gravidanza in donne che sono portatrici, anche se ogni caso è diverso e dovrebbe essere valutato individualmente nel contesto appropriato.
È stato notato che il rischio di presentare aritmia ventricolare maligna diminuisce con la gravidanza. Al contrario, è stata riportata una maggiore vulnerabilità a presentare un’aritmia maligna entro i primi 9 mesi dopo il parto, in particolare nei pazienti con LQTS2. Questo rischio diminuisce considerevolmente con la terapia beta-bloccante.96
STRATIFICAZIONE DEL RISCHIO
L’evoluzione della LQTS varia ed è influenzata dalla durata dell’intervallo QTc, da fattori ambientali, dall’età, dal genotipo e dalla risposta al trattamento.97,98 L’aritmia ventricolare è più frequente nella LQTS1 e LQTS2, ma è più grave nella LQTS3.99 Come è stato menzionato sopra, le donne sono particolarmente suscettibili all’aritmia maligna durante il periodo post-partum.14
La sindrome del QT lungo deve essere considerata ad alto rischio quando è associata a quanto segue:
1. Sordità congenita (sindrome di Jervell-Lange-Nielsen).
2. Sincope ricorrente dovuta a tachiaritmia ventricolare maligna.
3. Storia familiare di morte improvvisa.
4. QTc>500 ms.
5. Blocco atrioventricolare 2:1.
6. 6. Alternanza elettrica dell’onda T.
7. Genotipo LQTS3.
Lo studio di Priori et al97 eseguito su 647 pazienti ha dimostrato che la probabilità di presentare un evento maggiore (sincope, arresto cardiaco, morte improvvisa) prima dei 40 anni è alta (>50%) quando il QTc è >500 ms nella LQTS1, LQTS2, e nei maschi con LQTS3. Recentemente, è stata riportata un’analisi del registro internazionale LQTS. Il rischio di morte improvvisa è stato analizzato in 2772 adolescenti con la malattia, e sono stati identificati 3 fattori associati a un rischio maggiore in questa popolazione: QTc>530 ms, storia di sincope negli ultimi 10 anni e sesso; i ragazzi tra i 10 e i 12 anni avevano un rischio maggiore rispetto alle ragazze, ma nella fascia di età tra i 13 e i 20 anni il rischio era comparabile.100
TREATMENT
I pazienti sintomatici che non ricevono un trattamento hanno un tasso di mortalità annuale del 20% e una mortalità a 10 anni del 50% dopo un primo evento di aritmia ventricolare. Anche se è chiaro che il trattamento dovrebbe essere stabilito quando ci sono sintomi, l’approccio da usare nei pazienti asintomatici è ancora in discussione. È stato documentato che l’arresto cardiaco può essere la prima manifestazione della malattia nel 9% dei pazienti,48 e che il 12% dei pazienti asintomatici svilupperà dei sintomi e potrebbe andare incontro a morte improvvisa. Il trattamento iniziale con beta-bloccanti dovrebbe essere iniziato in tutti i pazienti con LQTS. La restrizione dell’esercizio fisico è raccomandabile, ma i marcatori di rischio clinico ed elettrocardiografico sono una base utile per il processo decisionale. È importante informare i pazienti sul rischio di usare diversi farmaci che possono prolungare l’intervallo QT e favorire lo sviluppo dell’aritmia ventricolare, come si è detto sopra. La diagnosi genetica, oltre a permettere un’adeguata consulenza familiare relativa alla malattia, è un aiuto per valutare la prognosi e orientare il trattamento specifico.
I beta-bloccanti
I beta-bloccanti sono il trattamento di prima linea per la LQTS e tutti i pazienti dovrebbero riceverli come terapia iniziale.101 Essi forniscono una riduzione del rischio di eventi cardiovascolari fino al 64%100 e sono particolarmente efficaci nei pazienti con mutazioni dei canali IKs (LQTS1),102 che sono regolati in larga misura dal sistema simpatico. I beta-bloccanti non modificano l’intervallo QT, ma la sua dispersione.103 Sebbene questi farmaci diminuiscano l’incidenza degli eventi104,105 è stato dimostrato che il 10% dei pazienti con LQTS1, il 23% con LQTS2 e il 32% con LQTS3 avranno sintomi cardiovascolari nonostante il trattamento.106 I pazienti con LQTS3, in particolare, non sembrano ottenere benefici importanti; infatti, questo gruppo di farmaci dovrebbe essere usato con cautela in questi pazienti, perché gli episodi di aritmia ventricolare nel LQTS3 sono più comuni quando la frequenza cardiaca è bassa. In termini generali, il 32% dei pazienti sintomatici avrà sintomi ricorrenti nei primi 5 anni prima di iniziare il trattamento con beta-bloccanti, e il 14% dei pazienti salvati da un episodio di morte improvvisa presenterà un altro evento simile entro 5 anni se riceve solo questa terapia.107 Diversi beta-bloccanti sono stati utilizzati nel trattamento della LQTS, principalmente nadololo (0,5-1 mg/kg/die), propranololo (2-4 mg/kg/die), metoprololo (0,5-1 mg/kg/die) e atenololo (0,5-1 mg/kg/die). L’atenololo potrebbe non essere benefico nella LQTS, tuttavia; è stato notificato che almeno il 75% dei pazienti che non hanno risposto alla terapia beta-bloccante stavano ricevendo atenololo, anche se questo risultato può essere legato all’uso di dosi subottimali.104 Il test da sforzo è utile per stabilire la dose appropriata. La frequenza cardiaca massima non dovrebbe superare i 130 battiti/min durante il trattamento.
Bloccanti del canale del sodio
Le mutazioni del canale del sodio che causano la LQTS3 producono un’inattivazione difettosa del canale; il blocco del canale del sodio si è dimostrato utile in questi pazienti. Gli studi condotti con la flecainide hanno documentato miglioramenti nella frequenza cardiaca, nelle alterazioni dell’onda T e nell’intervallo QT.108 È stato riportato che anche la mexiletina migliora i marcatori di rischio elettrocardiografici.63,109,110 Gli studi in vitro con la ranolazina hanno mostrato una diminuzione degli effetti deleteri delle mutazioni riportate nell’uomo.111 Sebbene i risultati siano incoraggianti, bisogna tenere presente che non ci sono studi a lungo termine che valutino questa terapia e non sono stati riportati risultati da grandi serie. I bloccanti dei canali del sodio non dovrebbero essere somministrati se non c’è una diagnosi genetica confermata.
Integrazione di potassio e farmaci che ne aumentano la disponibilità
Integratori di potassio e/o farmaci che risparmiano potassio, come lo spironolattone, accorciano l’intervallo QTc nel 24% dei casi.112,113 I farmaci che favoriscono l’apertura dei canali del potassio, come aprikalim, levcromakalim, nicorandil e pinacidil, hanno dimostrato di essere utili nel trattamento della LQTS. I sottotipi in cui sono di particolare beneficio sono LQTS1 e LQTS2.114
Pacemaker e defibrillatori
La stimolazione del pacemaker è stata utilizzata nei pazienti con aritmia dipendente dalla pausa.115,116 I pazienti con LQTS3 solitamente beneficiano maggiormente di questo trattamento perché la prevalenza di bradicardia è maggiore in questo gruppo. La stimolazione DDD è indicata nei pazienti con aritmia dipendente dalla pausa o blocco AV di alto grado 2:1. Le frequenze programmate sotto i 70 battiti/min117 non sono utili per prevenire l’aritmia ventricolare. Si raccomanda di programmare il sensore a risposta rapida, perché questi pazienti di solito hanno un’accelerazione inappropriata della frequenza cardiaca in risposta all’esercizio. Tutte le funzioni che implicano la presenza di pause dovrebbero essere disattivate, come l’isteresi e la funzione notturna. Il PARP (periodo refrattario atriale postventricolare) dovrebbe essere il più breve possibile. La funzione di regolazione della frequenza dovrebbe essere attivata per prevenire la pausa post-trasistolica. Si deve ricordare che anche l’oversensing dell’onda T e i fallimenti di cattura possono dare origine a pause. L’uso combinato di un defibrillatore cardioverter impiantabile (ICD) e di beta-bloccanti diminuisce sostanzialmente l’incidenza della morte improvvisa.118-120 L’indicazione per queste misure è chiara nei casi ad alto rischio.121 La programmazione del dispositivo varia a seconda delle esigenze del singolo paziente, ma, in generale, la somministrazione del trattamento in eventi asintomatici e autolimitati dovrebbe essere evitata; a tal fine, è indicato un tempo di rilevazione di 15 s. La tempesta aritmica è una complicazione della terapia AID. Quasi il 15% dei pazienti può sperimentare questa complicazione, che è dovuta, in buona parte, all’aumento del tono simpatico in seguito allo shock dell’ICD.118 Questo problema può essere gestito aumentando la dose di beta-bloccante. Se questa misura non è utile, si deve considerare la resezione dei gangli della catena simpatica.
Simpatectomia sinistra
Nel 1971 la gangliectomia simpatica è stata introdotta come un’opzione terapeutica utile in questi pazienti.122 Nel 1991, Schwartz et al123 hanno pubblicato la prima serie di 85 pazienti con una scarsa risposta al trattamento con beta-bloccanti, nei quali è stata eseguita una stellectomia sinistra con risultati incoraggianti: un tasso di sopravvivenza a 5 anni del 94%. Attualmente, questa opzione terapeutica viene offerta ai pazienti ad alto rischio che persistono con la sincope nonostante il trattamento beta-bloccante e/o l’impianto di pacemaker, e a quelli che subiscono frequenti shock dal loro defibrillatore impiantato. La procedura consiste nella resezione della porzione inferiore del ganglio stellato e dei gangli toracici sinistri da T2 a T4 della catena simpatica, poiché la semplice stellectomia sinistra non si è dimostrata sufficientemente efficace. La toracoscopia microinvasiva124,125 è stata utilizzata con buoni risultati. La più grande serie di pazienti trattati con questo metodo è stata recentemente riportata e ha mostrato una significativa riduzione del numero di episodi di sincope o di morti improvvise, nonché un tasso di sopravvivenza a 5 anni del 95%. Nei pazienti con precedenti sincopi, la sopravvivenza a 5 anni era del 97%, con una possibilità di recidiva dell’11%, che, nella maggioranza, consisteva in un singolo evento sincopale. C’era anche una significativa riduzione del segmento QT dopo la simpatectomia sinistra. Nonostante questi risultati favorevoli, la prevenzione della morte improvvisa non è completa, ma è stata ridotta al 3%. Nei pazienti con un ICD che si sono sottoposti all’intervento a causa di shock multipli del defibrillatore, il numero medio di eventi è diminuito da 25 a 0, una riduzione del 95%. Un effetto benefico è stato confermato nel LQTS1. È probabile che i benefici siano minori nei pazienti con LQTS2, e nel LQTS3 la sua efficacia non è stata dimostrata.126
Ablazione
È stato riportato che l’ablazione dell’extrasistole, che in alcuni casi inizia l’aritmia ventricolare, può essere effettuata con una riduzione dell’incidenza degli episodi.127 Tuttavia, non esistono studi a lungo termine con un numero adeguato di pazienti per giustificare l’uso di routine di questa tecnica.
Vedi editoriale alle pagine 675-82
ABBREVIAZIONI
AV: atrioventricolare
AID: defibrillatore automatico impiantabile
ECG: elettrocardiogramma
QTc: QT corretto per la frequenza cardiaca
ATS: Sindrome di Andersen-Tawil
LQTS: sindrome del QT lungo
Il Dr. Medeiros riceve supporto economico da CONACyT e FUNSALUD.