INTRODUKTION
Långt QT-syndrom (LQTS) kännetecknas av kraftigt förändrad ventrikulär repolarisering, vilket resulterar i en förlängning av QT-intervallet på elektrokardiogrammet (EKG). Tillståndet predisponerar patienter för malign ventrikulär arytmi (torsade de pointes) och plötslig död. Den kliniska och elektrokardiografiska beskrivningen av det långa QT-syndromet rapporterades 1957 av Anton Jervell och Fred Lange Nielsen,1 som publicerade sina studier på en familj av icke-konsanguina föräldrar med 6 barn. Fyra av barnen hade medfödd dövhet och synkopala episoder, och tre presenterade plötslig död. EKG-undersökning av dessa patienter visade ett ovanligt långt QT-intervall. Båda föräldrarna var symtomfria, hade ett normalt EKG och uppvisade inga hörselproblem. År 1964 rapporterade Romano och Ward oberoende av varandra ett hjärtsyndrom som kännetecknas av återkommande synkoper, en familjehistoria av plötslig död och förlängning av QT-intervallet utan neuronal dövhet.2 Senare genetiska studier visade att det syndrom som beskrivs av Jervell och Lange Nielsen, som åtföljs av medfödd neuronal dövhet, motsvarar homozygota mutationer, med en allvarlig fenotyp och hög risk för plötslig död. Det tillstånd som kallas Romano-Wards syndrom motsvarar i allmänhet heterozygota mutationer, patienterna uppvisar inga hörselförändringar och sjukdomens svårighetsgrad varierar avsevärt. Nästan ett halvt sekel senare, 19953,4 , beskrevs de viktigaste generna förknippade med LQTS och sjukdomen erkändes som en kardiell jonkanalsjukdom. Det var den första kardiella kanalopati som beskrevs och är kanske den mest utförligt undersökta arytmogena jonkanalsjukdomen hittills. Den kliniska bilden varierar kraftigt: patienten kan vara asymtomatisk eller uppvisa återkommande synkoper, kramper eller plötslig död som den första manifestationen av sjukdomen. Initialt betraktades LQTS som ett sällsynt syndrom och i själva verket är den allvarliga presentationen av sjukdomen sporadisk. Ändå uppskattas incidensen av relaterade mutationer till 1/3000-5000 fall,5 32 % av asymtomatiska bärare kan ha ett hjärtfrekvenskorrigerat QT-intervall (QTc) inom normala gränser, sjukdomen överförs till 50 % av deras ättlingar, de är mer mottagliga för att utveckla arytmi jämfört med befolkningen i allmänhet, och upp till 20 % kan få symtom.6
Långt QT-syndrom uppvisar stor genetisk heterogenitet. Mer än 500 mutationer fördelade på 10 gener har beskrivits i detta tillstånd: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 och SCN4B. Trots framstegen på detta område kan en genetisk diagnos inte fastställas hos 25-30 % av patienterna.7,8 Sjukdomsutbredningen är huvudsakligen monogen6; polygena eller sammansatta varianter har vanligen en allvarligare fenotyp. Penetranscensen, dvs. de patienter som har mutationen och uppvisar fenotypen, varierar mellan 25 % och 90 %.9 Mindre ofta kan det finnas variationer i sjukdomens uttrycksfullhet, med flera fenotyper som följd av samma mutation. Molekylärgenetiska studier som utvecklats under de senaste 11 åren har gett viktiga genotyp-fenotypkorrelationer, som har bidragit till att vägleda behandlingsmetoden. Dessutom har intressanta observationer gjorts om individuell känslighet för att utveckla arytmi i studier som undersöker de frekventa nonsynonyma polymorfismerna i denna population, en aspekt som har väckt stort intresse, särskilt inom området farmakogenomik.
KLASSIFICERING AV LONG QT SYNDROM
Allmänna begrepp
Den LQTS-klassificering som användes tidigare baserades på homozygot eller heterozygot uppkomst av sjukdomen, vilket ger upphov till Jervell-Lange-Nielsens syndrom (med dövhet) respektive Romano-Wards syndrom (utan dövhet). Den nuvarande klassificeringen betonar de genetiska fynden, vilket illustreras i tabell 1. De 3 viktigaste generna som är förknippade med sjukdomen beskrevs 1995-1996. Dessa gener, som kodar för porbildande enheter i kaliumkanalerna IKs och IKr samt natriumkanalen Nav1.5, står för nästan 65 % av fallen. Även om ytterligare sju gener under senare år har inkluderats i listan står de endast för 5 % av fallen.
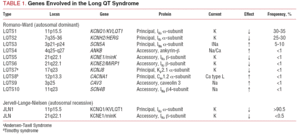
Ionkanaler är transmembranproteiner som transporterar joner genom cellmembranen. De kanaler som är inblandade i LQTS är selektiva eller specialiserade på att transportera en enda jon och är spänningsberoende, dvs. deras aktivering sker vid en specifik intracellulär spänning, som varierar beroende på kanalsubtyp. De elektriska och kontraktila fenomen som uppstår i kardiomyocyten styrs av dessa strukturer. Jonkanaler bildar makromolekylära komplex som består av en huvudenhet som bildar kanalporten och hjälpproteiner som reglerar den (figur 1). Den kanaldysfunktion som ses vid LQTS kan uppstå på dessa två ställen: huvudproteinet eller de reglerande proteinerna (tabell 1). Involvering av den porbildande enheten, känd som alfa, genererar de tre vanligaste subtyperna av LQTS: LQTS1 (som påverkar kaliumkanalen IKs), LQTS2 (som påverkar kaliumkanalen IKr) och LQTS3 (som påverkar natriumkanalen). Eftersom dessa är de vanligaste subtyperna är de bäst karakteriserade kliniskt och genetiskt. Fenotyp-genotypkorrelationerna i dessa tre huvudformer beskrivs i figur 2. För närvarande motsvarar Jervell-Lange-Nielsens syndrom LQTS 1- och 5varianterna. Karakteristiskt för dessa patienter är att de har medfödd dövhet och sammansatta homozygota eller heterozygota mutationer som påverkar IKs strömmen. Romano-Wards syndrom omfattar varianter från LQTS 1 till 10 och innebär inte dövhet.
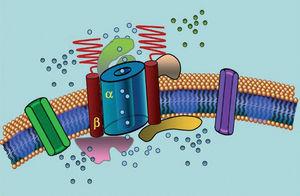
Figur 1. Schematisk bild av det makromolekylära komplexet. Jonkanalerna är transmembranproteiner (α) som regleras av olika proteiner; ett av dem är den så kallade β-underenheten.
Långt QT-syndrom typ 1 (LQTS1)
Patienter med LQTS1 uppvisar vanligen episoder av ventrikulär arytmi när de tränar eller när de utsätts för sympatisk stimulans (68 %).10 Simning har beskrivits som en sport som utlöser arytmi hos LQTS1.11 Penetranskänsligheten är nästan 62 % i denna subtyp. T-vågen hos dessa patienter har ofta en bred bas och mycket långvarig duration12,13 (figur 2). Det är den vanligaste subtypen och förklarar 30-35 % av fallen. Den drabbade genen, KvLQT1 (eller KCNQ1), ligger på kromosom 11 (11p15.5) och kodar för IKs-kaliumkanalens α-underenhet. Aktionspotentialen förlängs genom en minskning av den utgående K+-strömmen under aktionspotentialens fas 3.
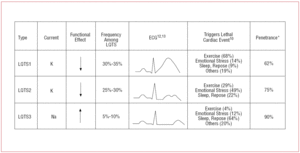
Figur 2. Genotyp-fenotypkorrelation i de vanligaste långa QT-syndromen. *Refekter avser fall som har mutationen och uppvisar fenotypen.
Långt QT-syndrom typ 2 (LQTS2)
Patienter med LQTS2 tenderar att uppvisa kammararytmi som svar på känslomässig stress (49 %) eller plötslig auditiv stimulans (t.ex. en väckarklocka), och mer sällan under sömn (22 %) eller träning (29 %).10 Kvinnor i postpartumperioden är särskilt känsliga.14 Den uppskattade penetransen är 79 %, vilket innebär att upp till 20 % av fallen kan ha ett icke-diagnostiskt EKG. T-vågen vid LQTS2 är vanligen lågamplitud och bifid, med notching12,13 (figur 2). Den drabbade genen är KCNH2 eller HERG, belägen på kromosom 7 (7q35-36), som kodar för IKr-kaliumkanalens α-underenhet och står för 25-30 % av fallen. Dysfunktion i denna kanal minskar den utgående K+-strömmen under aktionspotentialens fas 3, vilket förlänger dess varaktighet.
Långt QT-syndrom typ 3 (LQTS3)
Patienter med LQTS3 löper större risk att uppvisa maligna arytmier under vila (sömn) eller bradykardi.15 Penetranskännetecken för SCN5A-genmutationen är nästan 90 %. EKG vid LQTS3 visar vanligtvis en fördröjd, spetsig T-våg och gör det möjligt att tydligt observera ST-segmentförlängningen12,13 (figur 2). Dessa patienter har vanligtvis färre symtom än de med LQTS1 eller LQTS2, men händelserna är karakteristiskt sett mer dödliga.
Den drabbade genen i LQTS3 är SCN5A, som kodar för Nav1.5-natriumkanalens α-underenhet (figur 1), som är lokaliserad på kromosom 3 (3p21-24); den är orsaken till sjukdomen i 5-10 % av fallen. Defekt inaktivering av kanalen möjliggör en ihållande tillförsel av Na+ under aktionspotentialens fas 2, vilket förlänger dess varaktighet.
Långt QT-syndrom typ 4 (LQTS4)
Typ 4 är en sällsynt variant av LQTS och står för nästan 1 % av fallen. Det är en atypisk form som ger upphov till ett brett spektrum av arytmier, inklusive katekolaminerga polymorfa ventrikulära takykardi, förmaksflimmer, intraventrikulära konduktionsförändringar, sinusnodens dysfunktion och bradykardi6-18. Dessutom kan QTc ligga inom normala gränser hos många patienter. Den drabbade genen är ANKB, belägen på kromosom 4 (4q25-27), som kodar för syntesen av ankyrin-β, ett strukturellt protein som länkar kardiomyocytmembranproteiner till cytoskeletala proteiner. Dessa proteiner är Na/K ATPase-pumpen, Na/Ca-bytaren och inositoltrifosfatreceptorn (InsP3R). Mutationer som orsakar förlust av ankyrin-β-funktionen leder till ökningar av den intracellulära kalciumkoncentrationen och förändringar i uttrycket av N/K ATPas och Na/Ca-växlaren. Den förhöjda kalciumkoncentrationen ger upphov till tidiga och fördröjda efterdepolarisationer. De ventrikulära arytmier som observeras vid ankyrin-β-genmutationer beror således på spontana depolariseringar, vanligen som svar på katekolaminerga stimuleringar.
Långt QT-syndrom typ 5 (LQTS5)
Typ 5 har sitt ursprung i förändringar i sekvensen av KCNE1-genen som är belägen på kromosom 21 (21q22.1p22.)19 KCNE1 kodar för syntesen av IKs-kanalens β-underenhet, även känd som minK-underenheten, som reglerar IKs-kanalen. Denna typ står för mindre än 1 % av fallen.
Långt QT-syndrom typ 6 (LQTS6)
Den drabbade genen i typ 6 är KCNE2, belägen på kromosom 21 (21q22.1).20 Denna gen kodar för kaliumkanalens β-underenhet, även känd som MiRP1-underenheten, och den reglerar IKr-kanalen. Mindre än 1 % av fallen är av typ 6.
Långt QT-syndrom typ 7 eller Andersen-Tawils syndrom (LQTS7)
De dysmorfiska fynd och elektrokardiografiska förändringar som ses vid detta syndrom beskrevs första gången 1971 av dr Andersen21 och återbesöktes 1994 av dr Tawil22 , men den genetiska/molekylära beskrivningen rapporterades inte förrän 2001.23 Detta tillstånd, som nu kallas Andersen-Tawils syndrom (ATS), är en autosomalt dominant förändring som kännetecknas av periodisk förlamning, onormal skelettutveckling, ventrikulär arytmi av typen med frekventa ventrikulära extrasystoler och en särskild benägenhet att utveckla ventrikelflimmer, särskilt hos kvinnor. De förändringar som beskrivs i ATS omfattar ventrikulära extrasystoler (41 %), icke-underhållen polymorfisk ventrikulär takykardi (23 %), dubbelriktad ventrikulär takykardi (68 %) och torsade de pointes (3 %).24 Några av de observerade dysmorfiska egenskaperna är kortväxthet, skolios, klinodaktyli, hypertelorism, lågt placerade öron, mikrognathi och en bred panna. Sjukdomsuttrycket varierar, vilket försvårar en tidig diagnos.23,25 Mutationer i KCNJ2-genen som ligger på kromosom 17 (17q23) och som kodar för syntesen av den rektifierande kaliumkanalen Kir 2.1 står för 70 % av fallen. Denna kanal deltar i fas 4 av aktionspotentialen. Flera författare ifrågasätter inkluderandet av denna gen i LQTS-orsaksgruppen, eftersom QTc-intervallet endast är svagt förlängt vid detta syndrom eller till och med normalt, men U-vågen är vanligtvis framträdande, vilket har lett till en överskattning av QT-intervallet. Läsaren kommer att finna att vissa författare föreslår att KCNJ2-mutationer genererar ATS1 och inte LQTS7.24
Långt QT-syndrom typ 8 (LQTS8)
Typ 8 uppstår på grund av mutationer i CACNA1-genen som ligger på kromosom 12 (12p13.3) och som kodar för L-typ kalciumkanalen Cav1.2. Den orsakar Timothys syndrom,26 ett tillstånd som kännetecknas av hjärtmissbildningar, intermittent immunologisk brist, hypoglykemi, kognitiva förändringar inklusive autism, interdigital fusion och förlängd QT, vilket leder till hjärtarytmi och plötslig död.27 Mindre än 0,5 % av fallen är av typ 8.
Långt QT-syndrom typ 9 (LQTS9)
Denna variant av LQTS utvecklas av mutationer i CAV3-genen, belägen på kromosom 3 (3p25), som kodar för caveolin 3-syntes. Caveolin är en invagination av plasmamembranet som är involverad i endocytos, lipidhomeostas och signaltransduktion. En viktig komponent i denna struktur är caveolin, som har tre kända subtyper; subtyp 3 är specifik för skelett- och hjärtmuskulatur. Vissa jonkanaler är samlokaliserade i caveola, inklusive en kardiell isoform av natriumkanalen Nav1.5. Flera mutationer i detta protein har nyligen beskrivits. Dessa förändrar de biofysiska egenskaperna hos natriumkanalen Nav1.5 in vitro, vilket genererar en fenotyp som liknar den som observeras i LQTS3.28 Mindre än 1 % av fallen tillskrivs denna orsak.
Långt QT-syndrom typ 10 (LQTS10)
Typ 10 beskrevs i ett mycket allvarligt fall, med QTc >600 ms, fetal bradykardi och 2:1 atrioventrikulärt (AV) block. Den beror på mutationer i genen SCN4B, som ligger på kromosom 11 (11q23) och som kodar för natriumkanalens β4-underenhet. Fyra olika subtyper av β-subenheter har beskrivits, som interagerar och reglerar de olika isoformerna av natriumkanalen; trots detta har endast subtyp 4 hittills förknippats med arytmogenes.29 Incidensen av mutationer i denna gen har inte undersökts, men uppskattas till
Mutationer av Jervell-Lange-Nielsen-varianten
Denna allvarliga form av LQTS orsakas av homozygota30 eller sammansatta heterozygota mutationer i KCNQ1- och/eller KCNE1-genen, som kodar för IKs-strömmen; dvs. en mycket allvarlig variant av formerna LQTS1 eller LQTS5. Detta tillstånd är karakteristiskt förknippat med medfödd dövhet. Patienterna har vanligtvis en QTc>500 ms och återkommande synkoper och löper stor risk för plötslig död. Föräldrarna till patienter med denna variant är vanligen heterozygota och har mindre allvarlig sjukdom eller visar inga symtom.31
DIAGNOSIS AV LONG QT SYNDROM
Schwartz Score
År 1985 publicerade Schwartz et al32 kriterierna för att diagnostisera LQTS, som modifierades 1993 och som innehåller viktiga riktlinjer för den initiala utvärderingen av potentiella fall. I detta system används en poängsättning från 1 till 9 baserad på familjehistoria och de kliniska och elektrokardiografiska fynden. Sannolikheten för sjukdom är låg vid ≥1, medelhög vid 2-3 och hög vid ≥4 (tabell 2).

Prenataldiagnostik av långt QT-syndrom
Fosterbradykardi kan vara en av de första kliniska manifestationerna av LQTS. Retrospektiva serier har visat att upp till 70 % av de patienter som diagnostiserats med LQTS under barndomen har en historia av bradykardi, vanligtvis åtföljd av fetal hydrops.33 Bedömning av den fetala kardiella repolariseringen mellan vecka 14 och 39 är användbar för tidig diagnostik av LQTS.34
Gonadal mosaikism för LQTS har förknippats med återkommande fetala förluster under den tredje trimestern av graviditeten.35 Om sjukdomen är starkt misstänkt kan fostervattenprov efter 16 veckors graviditet vara användbart för att fastställa diagnosen, vilket är lätt att uppnå när en av föräldrarna är känd som bärare av en specifik mutation.36
STUDIE AV EN PATIENT MED LÅNGT QT-SYNDROM
Klinisk anamnes
En familje- och/eller personlig anamnes av plötslig död är av avgörande betydelse för både diagnos och riskstratifiering av LQTS. Dessutom kan utlösande faktorer och kontexten av synkope indikera LQTS-subtypen. Vid den inledande utvärderingen av ett misstänkt fall bör användning av läkemedel som kan förlänga QT-intervallet uteslutas.
QT-intervallet: Vad är normalt?
QT-intervallet bör företrädesvis mätas i ledningar II eller V5,37 där det har visat sig ha ett större prediktivt värde.38 Detta intervall anger varaktigheten av ventrikulär repolarisering och mäts från början av Q-vågen till slutet av T-vågen. Konventionellt används den formel som Bazett39 föreslog för att korrigera intervallets varaktighet i enlighet med hjärtfrekvensen (QTc=QT/√RR, uttryckt i sekunder). Även om det verkar enkelt att mäta QT-intervallet visste mindre än 40 % av andra läkare än kardiologer, mindre än 50 % av kardiologerna och mer än 80 % av specialisterna på arytmi i en multicenterstudie utförd av Viskin et al40 hur man mäter QT-intervallet på rätt sätt. Det är tillrådligt för läkare att utföra manuell mätning och inte lita på automatiserade mätningar, som kan vara användbara för andra intervall, men som är oprecisa vid beräkning av QT-intervallet. QT är ett dynamiskt intervall och de normala gränserna beror på flera faktorer. Även om ett QTc-intervall på é440 ms hos män och é460 ms hos kvinnor anses vara onormalt, kan man hitta bärare av mutationer såväl som friska individer inom detta intervall (figur 3). I familjer med LQTS1 visade Vincent et al41 att inget av fallen med positiv genotyp hade ett QTc470 ms. Monnig et al38 visade nyligen att QTc>440 ms räcker för att upptäcka patienter med LQTS-associerade mutationer, QTc>470 ms är användbart för att identifiera patienter som riskerar att utveckla symtom, och QTc>500 ms återfinns hos symtomatiska patienter som genomgår behandling.
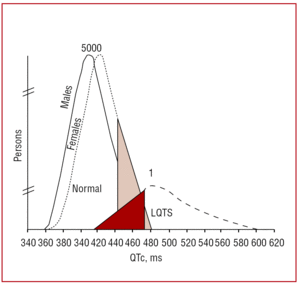
Figur 3. Modell som visar fördelningen av det hjärtfrekvenskorrigerade QT-intervallet (QTc) hos patienter med mutationer i KVLQT1, HERG eller SCN5A och deras opåverkade familjemedlemmar. Kurvan till vänster beskriver fördelningen av icke påverkade medlemmar och kurvan till höger, påverkade medlemmar.
Andra elektrokardiografiska förändringar associerade med långt QT-syndrom
Patienter med LQTS kan uppvisa flera T-vågsförändringar: polaritetsalterans, amplitudvariationer, notching och ett bifasiskt utseende, bland annat42. T-vågsalternering (figur 4A) definieras som en variation slag för slag i amplitud, morfologi och polaritet hos en T-våg i sinusrytm, utan variationer i QRS-komplexet. Det är en indikator på elektrisk instabilitet,43 som återspeglar regional spridning av ventrikulär repolarisering och föregår ibland ventrikelflimmer.44
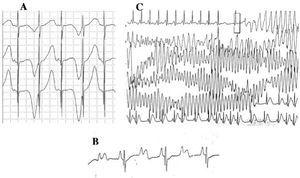
Figur 4. Elektrokardiografiska förändringar vid långt QT-syndrom. A: Elektrisk alternans av T-vågor. B: atrioventrikulärt block 2:1. C: Självbegränsad torsade de pointes.
Patienter med LQTS kan utvecklas med tecken på dysfunktion i sinusknutan, bradykardi och/eller pauser.45 Subtyperna LQTS1 och LQTS3, särskilt den senare, uppvisar ofta sinusbradykardi,46 medan LQTS4 har förknippats med sinusnodens dysfunktion.18
Sedan decenniet 1970-1980 har samexistens av AV-ledningsdefekter med LQTS47 observerats (figur 4B). Två till ett AV-block är en sällsynt manifestation med dålig prognos som kan förekomma sedan fosterstadiet i form av persisterande bradykardi. Incidensen av denna abnormitet har rapporterats till 4-5 %48 och är förknippad med hög mortalitet trots behandling med betablockerare och/eller pacemaker.49,50 Fenomenet kan förklaras av en långvarig duration av aktionspotentialen. När den ventrikulära refraktärperioden förlängs blockeras den följande impulsen av sinusaktivitet eftersom den når ventriklarna när de fortfarande befinner sig i refraktärperioden. Denna förändring tycks uteslutande inträffa vid LQTS, eftersom den ventrikulära refraktära perioden är längre än AV-ledningssystemets.51 QRS-komplexets lutning är vanligtvis brant och blocket har lokaliserats i infraHis-området,46,51,52 men platsen för blocket kan bero på genotypen. Hittills har 4 gener relaterats till 2:1-block i LQTS: HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 och SCN4B (LQTS10).55
Den karakteristiska ventrikulära arytmin för LQTS är känd som torsade de pointes (figur 4C). Den uppträder när QT-intervallet är förlängt, oavsett etiologi. Det är en polymorf ventrikulär takykardi på grund av reentry, som elektrokardiografiskt kännetecknas av kontinuerlig vridning av QRS-axeln runt en imaginär linje. Den föregås vanligen av en paus som följs av en extrasystole (kort-långt-kort RR-intervall), vilket visas i figuren.56-58 Den kan kulminera i kammarflimmer och plötslig död. Om detta inte inträffar kan patienten bara uppleva synkope, och om episoden är kortvarig kan den gå oupptäckt.
Holter
Holterstudie ger en fullständig, dynamisk bedömning av QT-intervallet. Ibland registreras spontana episoder av asymtomatisk ventrikulär arytmi, liksom episoder av sinusnodens dysfunktion eller AV-block.
Exercise Stress Test
Patienter med LQTS kan inte nå upp till den maximala förväntade hjärtfrekvensen beräknad enligt ålder. Dessutom kan QT-intervallet under ansträngning uppvisa ett paradoxalt beteende, genom att öka snarare än att minska.59,60 Det elektrokardiografiska mönstret under ansträngningstestning kommer att skilja sig åt beroende på typ av LQTS. Patienter med LQTS1 uppvisar, förutom att de inte når den maximala beräknade hjärtfrekvensen för sin ålder, ofta ett ökat QT-intervall, medan de med LQTS2 kan nå sin förväntade hjärtfrekvens och endast uppvisa en mild ökning av QT-intervallet eller ingen alls.61,62 Generellt sett har patienter med LQTS3 ett fysiologiskt svar på ansträngning, det vill säga normal förkortning av QT-intervallet.63 Stresstestning kan också vara användbart för att bedöma behandlingssvar och för att stratifiera risk i asymtomatiska fall, eller när det råder tvivel om de händelser som leder till arytmi.
Genetisk screening
Under de senaste åren har genetiska studier av LQTS varit begränsade till forskningslaboratorier. Trots detta har den information som härrör från dessa ansträngningar varit ytterst användbar för behandling av patienter, särskilt högriskfall. Den kanske viktigaste tillämpningen av screening är i genetisk rådgivning, men den har också viktiga implikationer i behandlingen, som kan orienteras enligt den drabbade kanalen. Den exakta placeringen av en viss mutation kan ge ytterligare information om riskutvecklingen. Patienter med mutationer i transmembranregionen hos KCNQ1 (IKS) har större sannolikhet att få arytmiska händelser än patienter med mutationer i den C-terminala regionen64; detsamma gäller för patienter med mutationer i porregionen hos KCNH2 eller HERG65 jämfört med patienter med mutationer i de N- eller C-terminala regionerna66 .
Inledande screening kan kanske begränsas till generna KCNQ1, HERG och SCN5A, som ger möjlighet att påträffa mutationer i 65 % av fallen. När de erhållna resultaten är negativa kan screeningen utökas till generna KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3 och SCN4B, vilket ökar möjligheten till positiva resultat med 5 % till 10 %.
Postmortal genetisk screening
Det är intressant att genmutationer som leder till LQTS har påträffats hos barn som upplevt plötslig död och i oförklarliga fall av plötslig död hos unga vuxna.
Genetiska undersökningar efter döden av patienter med plötslig död och negativ obduktion har visat mutationer som leder till LQTS i varierande procentandelar67-69: nära 10 % hos barn och 35 % hos unga vuxna.70-72 Baserat på dessa resultat har rutinmässig EKG-undersökning föreslagits hos alla nyfödda.73,74
Postmortala genetiska studier, som i litteraturen även kallas ”molekylär obduktion”, har förutom rättsliga återverkningar viktiga konsekvenser i familjer som kan påverkas utan att de vet om det.
Regulatoriska polymorfismer
Flera frekventa polymorfismer har beskrivits i LQTS-populationen, fördelade i nästan alla gener som är associerade med detta tillstånd. Även om dessa förändringar uppenbarligen inte är patogena kan vissa ha följande effekter75-78:
1. Generera individuell känslighet för att utveckla arytmi.
2. 2. Favoriserar den patogena effekten av en annan icke-synonym förändring.
3. Minskar den patogena effekten av en annan icke-synonym förändring.
Detta är fallet med K897T-polymorfismen i KCNH2 (HERG), som förekommer hos upp till 15 % av befolkningen och som inte bara är kopplad till känslighet för vissa läkemedel79 , utan som också gynnar den patogena effekten av mutationer i samma gen78. Ett annat exempel är S1103Y-polymorfismen i SCN5A-genen, som främst finns hos svarta, som har en förekomst på nästan 13 % och är förknippad med en ökad risk för plötslig död i barndomen80 .
Intressant nog har två alternativa bearbetningsställen som genererar två typer av natriumkanaler beskrivits i produkten av SCN5A-genen (som kodar för isoformen Nav1.5-natriumkanal hos människor): en med 2016 aminosyror som innehåller glutamin i position 1077 (Q1077) och en annan med 2015 aminosyror som saknar glutamin (Q1077del). Transkript av dessa alternativa bearbetningar finns i en 2:1-proportion i samma människohjärta och flera frekventa polymorfismer kommer att ha olika effekter på kanalens funktion, beroende på om kontexten är Q1077 eller Q1077del. Detta visades ursprungligen med polymorfismen H558R i SCN5A, som förekommer hos upp till 30 % av befolkningen. När H558R uttrycktes i samband med Q1077 observerades en kraftig minskning av jonströmmen.81 En liknande effekt dokumenterades med polymorfismen S524Y82. Dessa fynd har gett faktorer som förklarar sjukdomens varierande svårighetsgrad, liksom de olika fenotyper av samma mutation som observerats i vissa familjer.77
Farmakologisk testning med adrenalin
Farmakologisk testning med lågdos adrenalin är ett säkert och användbart alternativ för att avmaska misstänkta fall av LQTS med en gränslös QTc. Det är särskilt effektivt för att upptäcka asymtomatiska former av LQTS1, med en sensitivitet på 92,5 %, specificitet på 86 %, positivt prediktivt värde på 76 % och negativt prediktivt värde på 96 %. Det kan också vara användbart vid diagnos av LQTS2, med lägre sensitivitet och specificitet. Den är inte användbar för LQTS3 eller andra former av LQTS. Under normala förhållanden inducerar sympatisk stimulering fosforylering av kaliumkanalen IKs, vilket optimerar dess funktion och ger upphov till förkortning av aktionspotentialen. Hos patienter med LQTS, särskilt typ 1, observeras ett paradoxalt svar på administrering av lågdos adrenalin (0,025-0,2 µg/kg/min) som förlänger QT-intervallet till mer än 30 ms83-86.
QT-intervallförlängning och läkemedelsinducerad TORSADE DE POINTES
En stor mängd läkemedel som används inom olika medicinska specialiteter kan orsaka en iatrogen ökning av QT-intervallet. Vissa läkemedel har tagits bort från marknaden på grund av denna oönskade effekt (t.ex. astemizol och cisaprid, bland andra; mer information finns på www.qtdrugs.org).87,88
Ventrikulär arytmi som är sekundär till icke-antiarytmiska läkemedel förekommer hos mindre än en av 10 000 till 100 000 exponerade personer. Med tanke på att kliniska studier omfattar mellan 2 000 och 3 000 försökspersoner skulle denna oönskade och dödliga biverkning lätt undgå att upptäckas under den kliniska fasen av läkemedelsutvecklingen.89 Denna punkt har genererat ett enormt intresse för aspekter som rör säkerhet vid studier och utveckling av nya läkemedel.
De faktorer som är relaterade till individuell känslighet inkluderar kvinnligt kön, hypokalcemi, hypomagnesemi, bradykardi, hjärtsvikt, postkardioversion, förmaksflimmer, vänsterkammarhypertrofi, oupptäckt LQTS, predisponerande polymorfismer och höga serumkoncentrationer av predisponerande läkemedel.90
Den kanal som vanligtvis interagerar med läkemedel är IKr, som kodas av genen KCNH2(HERG), på grund av dess molekylära struktur. Andra kaliumkanaler har två prolinrester som är vinklade mot kanalporten, vilket minskar dess lumen. IKr saknar däremot dessa rester, en större porvestibul bildas och exponering av stora molekyler underlättas. Dessutom har den 2 aromatiska rester (tyrosin och fenylalanin) som gynnar bindning med aromatiska molekyler som finns i flera läkemedel som kan blockera kanalen.91
Som nämndes ovan är LQTS-penetrering ofullständig och vissa asymtomatiska bärare av mutationer kan manifestera malign arytmi efter att ha fått ett av dessa läkemedel. Dessutom ger polymorfismer som anses vara frekventa i befolkningen individuell känslighet för utveckling av torsade de pointes när vissa läkemedel används. Detta är fallet med R1047L-polymorfismen, den näst vanligaste i KCNH2, som har förknippats med utveckling av torsade de pointes vid användning av läkemedlet dofetilid.92 Minst 20 polymorfismer i KCNH2-genen har beskrivits hos friska personer och deras effekt på individens mottaglighet för att utveckla läkemedelsrelaterad arytmi återstår att fastställa.93 Polymorfismer som ger mottaglighet för utveckling av kammararytmi har också dokumenterats i natriumkanalen Na1.5. Detta gäller polymorfismen H558R, som förekommer hos upp till 30 % av befolkningen, eller S1103Y, som är vanlig hos svarta80,81,90,94,95; Deras betydelse för läkemedelsinducerad känslighet har inte undersökts.
LÅNG QT SYNDROM OCH PREGNANTI
Genetisk rådgivning är viktig vid LQTS, men generellt sett finns det ingen kontraindikation för graviditet hos kvinnor som är bärare av LQTS, även om varje fall är annorlunda och bör bedömas individuellt i ett lämpligt sammanhang.
Det har noterats att risken för att drabbas av maligna ventrikulära arytmier minskar med graviditet. Däremot har större sårbarhet för att presentera malign arytmi rapporterats inom de första 9 månaderna efter förlossningen, särskilt hos patienter med LQTS2. Denna risk minskar avsevärt vid behandling med betablockerare.96
RISKSTRATIFIERING
Utvecklingen av LQTS varierar och påverkas av QTc-intervallets varaktighet, miljöfaktorer, ålder, genotyp och respons på behandling.97,98 Ventrikulär arytmi är vanligare vid LQTS1 och LQTS2, men är allvarligare vid LQTS3.99 Som nämndes ovan är kvinnor särskilt mottagliga för malign arytmi under postpartumperioden.14
Långt QT-syndrom bör betraktas som högrisk när det är förknippat med följande:
1. Medfödd dövhet (Jervell-Lange-Nielsens syndrom).
2. Återkommande synkope på grund av malign ventrikulär takyarytmi.
3. Familjehistoria av plötslig död.
4. QTc>500 ms.
5. 2:1 atrioventrikulärt block.
6. T-vågs elektrisk alternans.
7. LQTS3-genotyp.
Studien av Priori et al97 som utfördes på 647 patienter visade att sannolikheten för att få en allvarlig händelse (synkope, hjärtstillestånd, plötslig död) före 40 års ålder är hög (>50 %) när QTc är >500 ms i LQTS1, LQTS2 och hos män med LQTS3. Nyligen rapporterades en analys av det internationella LQTS-registret. Risken för plötslig död analyserades hos 2772 ungdomar med sjukdomen, och 3 faktorer som är förknippade med högre risk i denna population identifierades: QTc>530 ms, historia av synkope under de senaste 10 åren och kön. 10-12-åriga pojkar hade en högre risk än flickor, men i åldersintervallet 13-20 år var risken jämförbar.100
BEHANDLING
Symtomatiska patienter som inte får någon behandling har en årlig dödlighet på 20 % och en 10-årsdödlighet på 50 % efter en första händelse av ventrikulär arytmi. Även om det står klart att behandling bör sättas in när det finns symtom, diskuteras fortfarande vilket tillvägagångssätt som ska användas hos asymtomatiska patienter. Det har dokumenterats att hjärtstillestånd kan vara den första manifestationen av sjukdomen hos 9 % av patienterna,48 och att 12 % av asymtomatiska patienter kommer att utveckla symtom och kan drabbas av plötslig död. Initial behandling med betablockerare bör inledas hos alla patienter med LQTS. Träningsbegränsning rekommenderas, men de kliniska och elektrokardiografiska riskmarkörerna är en användbar grund för beslutsfattandet. Det är viktigt att informera patienterna om risken med att använda flera läkemedel som kan förlänga QT-intervallet och gynna utvecklingen av ventrikulär arytmi, vilket nämns ovan. Genetisk diagnos är, förutom att möjliggöra lämplig familjerådgivning i samband med sjukdomen, en hjälp för att bedöma prognosen och orientera specifik behandling.
Betablockerare
Betablockerare är förstahandsbehandlingen vid LQTS och alla patienter bör få dem som första behandling.101 De ger en minskning av risken för kardiovaskulära händelser med upp till 64 %100 och är särskilt effektiva hos patienter med IKs-kanalmutationer (LQTS1),102 som i stor utsträckning regleras av det sympatiska systemet. Betablockerare ändrar inte QT-intervallet, utan i stället dess spridning.103 Även om dessa läkemedel minskar förekomsten av händelser104,105 har det visat sig att 10 % av patienterna med LQTS1, 23 % med LQTS2 och 32 % med LQTS3 kommer att ha kardiovaskulära symtom trots behandling106 . Särskilt patienter med LQTS3 verkar inte få några betydande fördelar; i själva verket bör denna läkemedelsgrupp användas med försiktighet hos dessa patienter, eftersom episoder av ventrikulär arytmi hos LQTS3 är vanligare när hjärtfrekvensen är låg. Generellt sett kommer 32 % av symptomatiska patienter att få återkommande symtom under de första 5 åren innan de påbörjar behandling med betablockerare, och 14 % av de patienter som räddas från en episod med plötslig död kommer att uppvisa en annan liknande händelse inom 5 år om de endast får denna behandling107 . Flera betablockerare har använts vid behandling av LQTS, främst nadolol (0,5-1 mg/kg/dag), propranolol (2-4 mg/kg/dag), metoprolol (0,5-1 mg/kg/dag) och atenolol (0,5-1 mg/kg/dag). Atenolol kanske dock inte är fördelaktigt vid LQTS; det har meddelats att minst 75 % av de patienter som inte svarade på behandling med betablockerare fick atenolol, även om detta resultat kan vara relaterat till användningen av suboptimala doser.104 Träningstestning är användbart för att fastställa lämplig dos. Maximal hjärtfrekvens bör inte överstiga 130 slag/min under behandlingen.
Natriumkanalblockerare
Natriumkanalmutationer som orsakar LQTS3 ger defekt inaktivering av kanalen; natriumkanalblockering har visat sig vara användbar hos dessa patienter. Studier som gjorts med flekainid har dokumenterat förbättringar av hjärtfrekvens, T-vågsförändringar och QT-intervall.108 Mexiletin har också rapporterats förbättra de elektrokardiografiska riskmarkörerna.63,109,110 In vitro-studier med ranolazin har visat minskningar av de skadliga effekterna av mutationer som rapporterats hos människor.111 Även om resultaten är uppmuntrande bör man komma ihåg att det inte finns några långtidsstudier som utvärderar denna behandling och inga rapporterade resultat från stora serier. Natriumkanalblockerare bör inte administreras om det inte finns någon bekräftad genetisk diagnos.
Kaliumtillskott och läkemedel som ökar dess tillgänglighet
Kaliumtillskott och/eller kaliumsparande läkemedel, t.ex. spironolakton, förkortar QTc-intervallet i 24 % av fallen112,113 . Läkemedel som gynnar öppning av kaliumkanalerna, såsom aprikalim, levcromakalim, nicorandil och pinacidil, har visat sig vara användbara vid behandling av LQTS. De subtyper där de är särskilt fördelaktiga är LQTS1 och LQTS2.114
Pacemakers och defibrillatorer
Stimulering av pacemaker har använts hos patienter med pausberoende arytmi.115,116 Patienter med LQTS3 har vanligen större nytta av denna behandling eftersom prevalensen av bradykardi är större i denna grupp. DDD-stimulering är indicerad hos patienter med pausberoende arytmi eller höggradigt 2:1 AV-block. Frekvenser programmerade under 70 slag/min117 är inte användbara för att förebygga ventrikulär arytmi. Det rekommenderas att programmera sensorn till snabb respons, eftersom dessa patienter vanligtvis har olämplig hjärtfrekvensacceleration som svar på träning. Alla funktioner som innebär närvaro av pauser bör stängas av, t.ex. hysteresis- och nattfunktion. PARP (postventrikulär atriell refraktärperiod) bör vara så kort som möjligt. Frekvensregleringsfunktionen bör vara påslagen för att förhindra postextrasystolisk paus. Man bör komma ihåg att T-vågsövervakning och fel i fångsten också kan ge upphov till pauser. Kombinerad användning av en implanterbar kardioverterdefibrillator (ICD) och betablockerare minskar väsentligt förekomsten av plötslig död.118-120 Indikationen för dessa åtgärder är tydlig i högriskfall.121 Programmeringen av anordningen kommer att variera beroende på den enskilda patientens behov, men generellt sett bör administrering av behandling vid asymtomatiska, självbegränsade händelser undvikas; för detta ändamål är en detektionstid på 15 s indicerad. Arytmisk storm är en komplikation vid AID-behandling. Nästan 15 % av patienterna kan drabbas av denna komplikation, som till stor del beror på ökad sympatikustonus efter ICD-chocken.118 Detta problem kan hanteras genom att öka dosen betablockerare. Om denna åtgärd inte är användbar bör resektion av den sympatiska kedjans ganglier övervägas.
Vänster sympathektomi
I 1971 introducerades sympathetisk gangliektomi som ett användbart terapeutiskt alternativ för dessa patienter.122 1991 publicerade Schwartz et al123 den första serien med 85 patienter som svarade dåligt på behandling med betablockerare och hos vilka man utförde en vänstersympathektomi med uppmuntrande resultat: en femårig överlevnadsfrekvens på 94 %. För närvarande erbjuds detta behandlingsalternativ till högriskpatienter som fortsätter att drabbas av synkope trots behandling med betablockerare och/eller pacemakerimplantation, och till dem som ofta får stötar från sin implanterade defibrillator. Förfarandet består av en resektion av den nedre delen av stellatanglionet och vänster thorakala ganglier T2 till T4 i den sympatiska kedjan, eftersom enkel vänster stellektomi inte har visat sig vara tillräckligt effektiv. Mikroinvasiv thorakoskopi124,125 har använts med goda resultat. Den största serien av patienter som behandlats med denna metod rapporterades nyligen och visade en betydande minskning av antalet synkopeepisoder eller plötsliga dödsfall samt en 5-årsöverlevnad på 95 %. Hos patienter med tidigare synkoper var 5-årsöverlevnaden 97 %, med 11 % risk för återfall, som i de flesta fall bestod av en enda synkopehändelse. Det fanns också en signifikant minskning av QT-segmentet efter vänster sympatektomi. Trots dessa gynnsamma resultat är förebyggandet av plötslig död inte fullständigt, men har minskat till 3 %. Hos patienter med en ICD som genomgick operation på grund av flera defibrillatorstötar minskade det genomsnittliga antalet händelser från 25 till 0, en 95-procentig minskning. En gynnsam effekt bekräftades för LQTS1. Fördelarna är sannolikt mindre hos patienter med LQTS2, och hos LQTS3 har dess effektivitet inte bevisats.126
Ablation
Det har rapporterats att ablation av extrasystolen, som i vissa fall initierar den ventrikulära arytmin, kan utföras med minskad förekomst av episoder.127 Det finns dock inga långtidsstudier med ett lämpligt antal patienter för att motivera rutinmässig användning av denna teknik.
Se ledare på sidorna 675-82
FÖRKORTNINGAR
AV: atrioventrikulär
AID: automatisk implanterbar defibrillator
ECG: elektrokardiogram
QTc: hjärtrytmekorrigerad QT
ATS: Andersen-Tawils syndrom
LQTS: långt QT-syndrom
Dr Medeiros får ekonomiskt stöd från CONACyT och FUNSALUD.