El término braquidactilia engloba un grupo de displasias óseas que afectan a las falanges y/o a los huesos metacarpianos/metatarsianos de las manos y los pies. Existen 5 tipos (A-E) y varios subtipos (A1-A4; E1-E3). Tiene un patrón de herencia autosómico dominante con penetrancia variable. Los casos con herencia recesiva son extremadamente raros.
La braquidactilia tipo C (BDC) se caracteriza por un acortamiento de las falanges medias del segundo, tercer y quinto dedo y del primer metacarpiano, y también puede presentar una desviación cubital del dedo índice, polidactilia o una hipersegmentación distintiva de las falanges proximales o medias del segundo y tercer dedo. El cuarto dedo es el menos afectado y suele ser el más largo.1-3 Las falanges «en forma de ángel» (Fig. 1A), aunque son características de la CDB, no son patognomónicas, ya que también pueden darse en el trastorno conocido como displasia falango-epifisaria en forma de ángel. Es posible que este trastorno y el CDB formen parte del mismo espectro clínico.1 En el CDB, este rasgo se normaliza cuando se completa el cierre fisiológico de los huesos de la mano, terminando como braquidactilia simple.1 Otras anomalías asociadas al CDB son la baja estatura con retraso de la edad ósea, la deformidad de Madelung, la displasia de cadera, el talipes valgo o equinovaro o la ausencia de falanges medias en los dedos de los pies.2,3
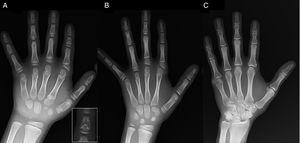
(A) Radiografía de la mano izquierda del probando (edad cronológica, 7 años; edad ósea, 5 años), con acortamiento de un primer metacarpiano anómalo (doble placa epifisaria proximal y distal) y de las falanges medias del segundo, tercer y quinto dedos. El segundo dedo presenta desviación cubital, y el cuarto dedo es el menos afectado y el más largo de la mano izquierda. Las epífisis proximales del segundo y tercer dedo son displásicas, con una falange media conspicua en forma de ángel en el segundo dedo (recuadro en A). (B). Radiografía de la mano izquierda de la hermana del probando (de 5½ años de edad sin retraso en la edad ósea), con acortamiento de las falanges medias de los dedos segundo, tercero y quinto y un cuarto dedo normal. En este caso, el primer metacarpiano era normal. La característica más destacada es la forma triangular de la epífisis proximal de la falange proximal del segundo dedo, similar a la del hermano, y la forma trapezoidal de la falange en el buscador medio. Al igual que su hermano, el segundo dedo presenta desviación cubital. (C) Radiografía de la mano izquierda del padre del probando, que revela sólo un remanente óseo de una hexadactilia post-axial que fue corregida quirúrgicamente en la infancia.
Presentamos el caso de un niño de 7 años de edad con características radiológicas compatibles con CDB remitido para evaluación de baja estatura (puntuación z, -1,8). Su padre tenía polidactilia post-axial unilateral, que también estaba presente bilateralmente en un tío paterno. El examen radiográfico de la mano y la muñeca izquierdas reveló una edad ósea 2 años inferior a la edad cronológica y anomalías que motivaron la realización de un estudio esquelético. El estudio detectó anomalías en las manos (Fig. 1A) y sutiles anomalías en los pies (displasia epifisaria leve en las falanges proximales de algunos dedos). La evaluación radiológica de una hermana de 6 años reveló lesiones en la mano similares a las del probando, aunque menos pronunciadas (Fig. 1B). Una radiografía de la mano del padre (Fig. 1C) sólo reveló un remanente óseo de la hexadactilia post-axial que había sido corregida quirúrgicamente en la infancia.
La secuenciación del gen GDF5 en el probando reveló una sustitución puntual heterocigótica en el exón 2 (c.1462A>T) que da lugar a un codón de parada prematuro (p.Lys488*, mutación sin sentido) y a una proteína truncada 14 aminoácidos más corta que la proteína salvaje (Fig. 2A). Esta mutación también se encontró en el padre y la hermana del paciente, pero no en la madre sana (Fig. 2B). La Fig. 2C muestra el pedigrí de la familia. Se trata de una nueva variante probablemente patogénica y con un patrón de herencia autosómico dominante.
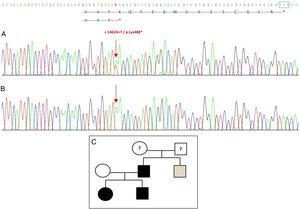
Nueva mutación detectada en el gen GDF5. (A) Secuencia génica del exón 2 en el probando, que muestra una mutación c.1462A>T que da lugar a un codón de parada prematuro y a una proteína truncada (p.Lys488*) (hemos indicado la posición normal del codón de parada con un cuadrado verde). (B) Secuencia normal de la misma región en la madre sana. (C) Pedigrí de la familia: el padre y los dos hijos, en negro, tienen una mutación confirmada en GDF5. La madre sana aparece en blanco. El cuadrado gris representa a un tío paterno con polidactilia bilateral post-axial que probablemente tiene la mutación.
El factor de diferenciación del crecimiento 5 (GDF5) está estrechamente relacionado con las proteínas morfogenéticas óseas y pertenece a la superfamilia del factor de crecimiento transformante β, con está implicado en el desarrollo embrionario del esqueleto y las articulaciones.4 El gen GDF5 es un punto caliente de mutaciones para los trastornos asociados a las malformaciones esqueléticas.5 La mayoría de las mutaciones homocigóticas o heterocigóticas compuestas están asociadas a enfermedades graves: Condrodisplasia tipo Grebe (OMIM 200700), displasia acromesomélica tipo Hunter-Thomson (OMIM 201250) o síndrome de Du Pan (OMIM 228900). Por otro lado, las mutaciones heterocigotas asociadas a displasias esqueléticas más leves: el sinfalangismo proximal 1 B (OMIM 615298) y el síndrome de sinostosis múltiple tipo 2 (OMIM 610017), ambos asociados a mutaciones sin sentido con ganancia de función, y la braquidactilia tipo A1 y A2, también asociadas a mutaciones sin sentido, pero con pérdida de función.5 La braquidactilia tipo C está asociada a mutaciones heterocigotas con pérdida de función, aunque también se han descrito 3 casos con herencia recesiva.6 La mayoría de las mutaciones asociadas a la CDB son mutaciones de desplazamiento de marco en la parte del pro-dominio del gen, mientras que la mayoría de las mutaciones en el dominio maduro son mutaciones sin sentido, con una expresión fenotípica muy variable.4
La familia que presentamos aquí tiene una mutación sin sentido en la región que codifica para el dominio activo de la proteína, lo que resulta en la eliminación de sus últimos 14 aminoácidos. Esta es la segunda mutación sin sentido que afecta al dominio activo maduro descrita en la literatura.3 La primera es una mutación similar en el aminoácido inmediatamente anterior al mutado en la familia que describimos aquí (p.Tyr487*/c.1461T>G), lo que sugiere que ambas dan lugar a monómeros mutantes y a la haploinsuficiencia funcional de GDF5, causando así la CDB.
Financiación
Este estudio ha sido parcialmente financiado por los proyectos PI13/00467 y PI13/01295, integrados en el plan R&D&I del Gobierno de España y cofinanciado por la Subdirección General de Evaluación y Fomento de la Investigación del Instituto de Salud Carlos III (ISCIII), el Fondo Europeo de Desarrollo Regional (FEDER) y el Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición, ISCIII, Madrid.