Der Begriff Brachydaktylie umfasst eine Gruppe von Knochendysplasien, die die Phalangen und/oder Mittelhand-/Metatarsalknochen der Hände und Füße betreffen. Es gibt 5 Typen (A-E) und mehrere Untertypen (A1-A4; E1-E3). Es handelt sich um einen autosomal dominanten Erbgang mit unterschiedlicher Penetranz. Fälle mit rezessivem Erbgang sind extrem selten.
Die Brachydaktylie Typ C (BDC) ist durch eine Verkürzung der Mittelglieder des zweiten, dritten und fünften Fingers und des ersten Mittelhandknochens gekennzeichnet und kann auch mit einer Ulnardeviation des Zeigefingers, Polydaktylie oder einer ausgeprägten Hypersegmentierung der proximalen oder mittleren Glieder des zweiten und dritten Fingers einhergehen. Der vierte Finger ist am wenigsten betroffen und in der Regel am längsten.1-3 „Engelsförmige“ Phalangen (Abb. 1A) sind zwar charakteristisch für BDC, aber nicht pathognomonisch, da sie auch bei der als engelsförmige Phalango-epiphysäre Dysplasie bekannten Störung auftreten können. Es ist möglich, dass diese Störung und die BDC Teil desselben klinischen Spektrums sind.1 Bei der BDC normalisiert sich dieses Merkmal, wenn der physeale Verschluss der Handknochen abgeschlossen ist, und endet als einfache Brachydaktylie.1 Andere Anomalien, die mit der BDC assoziiert sind, sind Kleinwuchs mit verzögertem Knochenalter, Madelung-Deformität, Hüftdysplasie, Talipes valgus oder equinovarus oder das Fehlen von Mittelphalangen an den Zehen.2,3
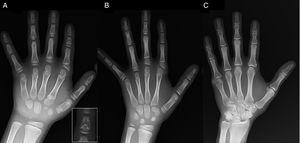
(A) Röntgenbild der linken Hand des Probanden (chronologisches Alter, 7 Jahre; Knochenalter, 5 Jahre), mit Verkürzung eines anomalen ersten Mittelhandknochens (doppelte proximale und distale Epiphysenplatten) und der Mittelphalangen des zweiten, dritten und fünften Fingers. Der zweite Finger weist eine Ulnardeviation auf, und der vierte Finger ist am wenigsten betroffen und an der linken Hand am längsten. Die proximalen Epiphysen des zweiten und dritten Fingers sind dysplastisch, mit einer auffälligen engelsförmigen Mittelphalanx am zweiten Finger (Inset in A). (B). Röntgenbild der linken Hand der Schwester des Probanden (im Alter von 5½ Jahren, ohne Verzögerung des Knochenalters), mit Verkürzung der Mittelphalangen des zweiten, dritten und fünften Fingers und einem normalen vierten Finger. In diesem Fall war der erste Mittelhandknochen normal. Das auffälligste Merkmal ist die dreieckige Form der proximalen Epiphyse des proximalen Fingerglieds des zweiten Fingers, die der des Bruders ähnelt, und die trapezförmige Form des Fingerglieds im Mittelfinger. Wie ihr Bruder weist auch ihr zweiter Finger eine Ulnardeviation auf. (C) Röntgenbild der linken Hand des Vaters der Probandin, das nur einen knöchernen Rest einer postaxialen Hexadaktylie zeigt, die in der Kindheit chirurgisch korrigiert wurde.
Wir stellen den Fall eines Jungen im Alter von 7 Jahren mit radiologischen Merkmalen vor, die mit einer BDC vereinbar sind, der zur Beurteilung seines Kleinwuchses (z-score, -1,8) überwiesen wurde. Sein Vater hatte eine einseitige post-axiale Polydaktylie, die auch bei einem Onkel väterlicherseits beidseitig vorhanden war. Die Röntgenuntersuchung der linken Hand und des linken Handgelenks ergab ein um 2 Jahre jüngeres Knochenalter als das chronologische Alter und Anomalien, die eine Skelettuntersuchung erforderlich machten. Bei der Untersuchung wurden Anomalien an den Händen (Abb. 1A) und subtile Anomalien an den Füßen (leichte epiphysäre Dysplasie an den proximalen Phalangen einiger Zehen) festgestellt. Die radiologische Untersuchung einer Schwester im Alter von 6 Jahren ergab ähnliche Läsionen an der Hand wie bei der Probandin, wenn auch weniger ausgeprägt (Abb. 1B). Eine Röntgenaufnahme der Hand des Vaters (Abb. 1C) zeigte nur einen Knochenrest der postaxialen Hexadaktylie, die in der Kindheit chirurgisch korrigiert worden war.
Die Sequenzierung des GDF5-Gens bei der Probandin ergab eine heterozygote Punktsubstitution in Exon 2 (c.1462A>T), die zu einem vorzeitigen Stoppcodon (p.Lys488*, Nonsense-Mutation) und einem verkürzten Protein führt, das 14 Aminosäuren kürzer ist als das Wildprotein (Abb. 2A). Diese Mutation wurde auch beim Vater und der Schwester der Patientin gefunden, nicht aber bei der gesunden Mutter (Abb. 2B). Abb. 2C zeigt den Stammbaum der Familie. Es handelt sich um eine neue Variante, die wahrscheinlich pathogen ist und einen autosomal dominanten Erbgang aufweist.
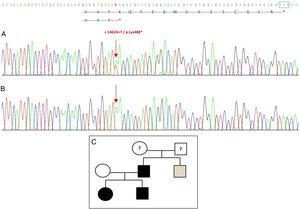
Neue Mutation im GDF5-Gen entdeckt. (A) Gensequenz von Exon 2 bei der Probandin, die eine c.1462A>T-Mutation zeigt, die zu einem vorzeitigen Stoppcodon und einem verkürzten Protein (p.Lys488*) führt (die normale Position des Stoppcodons ist mit einem grünen Quadrat gekennzeichnet). (B) Normale Sequenz der gleichen Region bei der gesunden Mutter. (C) Stammbaum der Familie: der Vater und beide Kinder, in schwarz, haben eine bestätigte Mutation in GDF5. Die gesunde Mutter erscheint in weiß. Das graue Quadrat stellt einen Onkel väterlicherseits mit bilateraler postaxialer Polydaktylie dar, der wahrscheinlich die Mutation hat.
Der Wachstumsdifferenzierungsfaktor 5 (GDF5) ist eng mit den knochenmorphogenetischen Proteinen verbunden und gehört zur Superfamilie der transformierenden Wachstumsfaktoren β, die an der embryonalen Skelett- und Gelenkentwicklung beteiligt sind.4 Das GDF5-Gen ist ein Mutations-Hotspot für Erkrankungen, die mit Skelettfehlbildungen einhergehen.5 Die meisten homozygoten oder compound heterozygoten Mutationen sind mit schweren Erkrankungen verbunden: Chondrodysplasie vom Typ Grebe (OMIM 200700), akromesomelische Dysplasie vom Typ Hunter-Thomson (OMIM 201250) oder Du-Pan-Syndrom (OMIM 228900). Andererseits werden heterozygote Mutationen mit milderen Skelettdysplasien in Verbindung gebracht: Proximaler Symphalangismus 1 B (OMIM 615298) und Multiples Synostose-Syndrom Typ 2 (OMIM 610017), die beide mit Missense-Mutationen mit Funktionsgewinn assoziiert sind, sowie Brachydaktylie Typ A1 und A2, die ebenfalls mit Missense-Mutationen, aber mit Funktionsverlust assoziiert sind.5 Brachydaktylie Typ C wird mit heterozygoten Mutationen mit Funktionsverlust assoziiert, obwohl auch 3 Fälle mit rezessivem Erbgang berichtet wurden.6 Die meisten Mutationen, die mit BDC assoziiert sind, sind Frameshift-Mutationen im Prodomain-Teil des Gens, während die meisten Mutationen in der reifen Domäne Missense-Mutationen sind, mit einer sehr variablen phänotypischen Ausprägung.4
Die Familie, die wir hier vorstellen, hat eine Nonsense-Mutation in der Region, die für die aktive Domäne des Proteins kodiert, was zur Eliminierung der letzten 14 Aminosäuren führt. Dies ist die zweite in der Literatur beschriebene Nonsense-Mutation, die die aktive reife Domäne betrifft.3 Bei der ersten handelt es sich um eine ähnliche Mutation in der Aminosäure, die der in der hier beschriebenen Familie mutierten unmittelbar vorausgeht (p.Tyr487*/c.1461T>G), was darauf hindeutet, dass beide zu mutierten Monomeren und funktioneller Haploinsuffizienz von GDF5 führen und damit BDC verursachen.
Finanzierung
Diese Studie wurde teilweise durch die Projekte PI13/00467 und PI13/01295 finanziert, die in den Plan R&D&I der spanischen Regierung für den Zeitraum 2013-2016 integriert sind und von der stellvertretenden Generaldirektion für Forschungsbewertung und -förderung des Instituto de Salud Carlos III (ISCIII) mitfinanziert werden, dem Europäischen Fonds für regionale Entwicklung (EFRE) und dem Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición (Biomedical Research Networking Centre on the Physiopathology of Obesidad and Nutrition ), ISCIII, Madrid.