De term brachydactylie omvat een groep botdysplasieën waarbij de vingerkootjes en/of middenhandsbeentjes/metatarsale botjes van de handen en voeten betrokken zijn. Er zijn 5 types (A-E) en verschillende subtypes (A1-A4; E1-E3). Het heeft een autosomaal dominant overervingspatroon met variabele penetrantie. Gevallen met recessieve overerving zijn uiterst zeldzaam.
Brachydactylie type C (BDC) wordt gekenmerkt door een verkorting van de middelste vingerkootjes van de tweede, derde en vijfde vinger en het eerste middenhandsbeentje, en kan ook gepaard gaan met ulnaire deviatie van de wijsvinger, polydactylie, of een opvallende hypersegmentatie van de proximale of middelste vingerkootjes van de tweede en derde vinger. De vierde vinger is het minst aangetast en meestal het langst.1-3 “Engelvormige” vingerkootjes (fig. 1A) zijn weliswaar kenmerkend voor BDC, maar zijn niet pathognomonisch, omdat zij ook kunnen voorkomen bij de aandoening die bekend staat als engelvormige falango-epiphyseale dysplasie. Het is mogelijk dat deze aandoening en BDC deel uitmaken van hetzelfde klinische spectrum.1 Bij BDC normaliseert dit kenmerk wanneer de fyseale sluiting van de handbeentjes is voltooid, eindigend als eenvoudige brachydactylie.1 Andere afwijkingen die in verband worden gebracht met BDC zijn een korte gestalte met vertraagde botleeftijd, Madelung misvorming, heupdysplasie, talipes valgus of equinovarus of afwezigheid van middenkootjes in de tenen.2,3
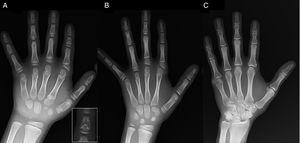
(A) Röntgenfoto van de linkerhand van de proband (chronologische leeftijd, 7 jaar; botleeftijd, 5 jaar), met verkorting van een afwijkend eerste middenhandsbeentje (dubbele proximale en distale epifysaire platen) en de middelste vingerkootjes van de tweede, derde en vijfde vinger. De tweede vinger vertoont ulnaire deviatie, en de vierde vinger is het minst aangetast en het langst in de linkerhand. De proximale epifysen van de tweede en derde vinger zijn dysplastisch, met een opvallende engelvormige middelste falanx in de tweede vinger (inzet in A). (B). Röntgenfoto van de linkerhand van de zus van de proband (5½ jaar oud zonder vertraging in de botleeftijd), met verkorting van de middelste vingerkootjes van de tweede, derde en vijfde vinger en een normale vierde vinger. In dit geval was het eerste middenhandsbeentje normaal. Het meest opvallende kenmerk is de driehoekige vorm van de proximale epifyse van de proximale falanx van de tweede vinger, vergelijkbaar met die van de broer, en de trapeziumvormige vorm van het vingerkootje in de middelste vingerkoot. Net als bij haar broer, vertoont haar tweede vinger ulnaire deviatie. (C) Röntgenfoto van de linkerhand van de vader van de proband, die alleen een benig overblijfsel toont van een postaxiale hexadactylie die chirurgisch werd gecorrigeerd in de kindertijd.
We presenteren het geval van een jongen van 7 jaar met radiologische kenmerken die compatibel zijn met BDC die werd doorverwezen voor evaluatie van een klein gestalte (z-score, -1,8). Zijn vader had unilaterale post-axiale polydactylie, die ook bilateraal aanwezig was bij een oom van vaderszijde. Radiografisch onderzoek van de linkerhand en pols toonde een botleeftijd die 2 jaar jonger was dan de chronologische leeftijd en anomalieën die aanleiding gaven tot het uitvoeren van een skeletonderzoek. Het onderzoek toonde afwijkingen aan in de handen (Fig. 1A) en subtiele afwijkingen in de voeten (milde epiphyseale dysplasie in de proximale vingerkootjes van sommige tenen). De radiologische evaluatie van een zusje van 6 jaar toonde laesies in de hand vergelijkbaar met die van de proband, hoewel minder uitgesproken (Fig. 1B). Een röntgenfoto van de hand van de vader (Fig. 1C) toonde alleen een botrestant van de post-axiale hexadactylie die in de kindertijd operatief was gecorrigeerd.
Sequencing van het GDF5-gen bij de proband toonde een heterozygote puntvervanging in exon 2 (c.1462A>T), resulterend in een voortijdig stopcodon (p.Lys488*, nonsens mutatie) en een afgekapt eiwit dat 14 aminozuren korter is dan het wilde eiwit (Fig. 2A). Deze mutatie werd ook gevonden in de vader en zuster van de patiënte, maar niet in de gezonde moeder (Fig. 2B). Fig. 2C toont de stamboom van de familie. Dit is een nieuwe variant die waarschijnlijk pathogeen is en met een autosomaal dominant overervingspatroon.
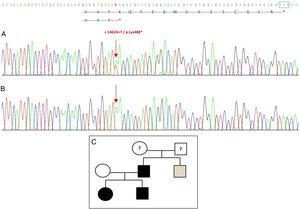
Nieuwe mutatie ontdekt in het GDF5-gen. (A) Gensequentie van exon 2 in de proband, met een c.1462A>T mutatie die resulteert in een voortijdig stopcodon en een afgekapt eiwit (p.Lys488*) (we hebben de normale positie van het stopcodon aangegeven met een groen vierkantje). (B) Normale sequentie van dezelfde regio in de gezonde moeder. (C) Stamboom van het gezin: de vader en beide kinderen, in zwart, hebben een bevestigde mutatie in GDF5. De gezonde moeder verschijnt in het wit. Het grijze vierkant vertegenwoordigt een oom van vaderskant met bilaterale post-axiale polydactylie die waarschijnlijk de mutatie heeft.
Groei differentiatiefactor 5 (GDF5) is nauw verbonden met botmorfogenetische eiwitten en behoort tot de transformerende groeifactor β superfamilie, met is betrokken bij de embryonale skelet- en gewrichtsontwikkeling.4 Het GDF5 gen is een mutatie-hotspot voor aandoeningen die geassocieerd worden met skeletmisvormingen.5 De meeste homozygote of samengestelde heterozygote mutaties worden geassocieerd met ernstige ziekten: Grebe type chondrodysplasie (OMIM 200700), Hunter-Thomson type acromesomelische dysplasie (OMIM 201250) of Du Pan syndroom (OMIM 228900). Anderzijds zijn heterozygote mutaties geassocieerd met mildere skeletdysplasieën: proximale symphalangie 1 B (OMIM 615298) en multiple synostose syndroom type 2 (OMIM 610017), beide geassocieerd met missense mutaties met functievermeerdering, en brachydactylie type A1 en A2, ook geassocieerd met missense mutaties, maar met functieverlies.5 Brachydactylie type C is geassocieerd met heterozygote mutaties met functieverlies, hoewel er ook 3 gevallen met recessieve overerving zijn gerapporteerd.6 De meeste mutaties geassocieerd met BDC zijn frameshift mutaties in het pro-domein deel van het gen, terwijl de meeste mutaties in het rijpe domein missense mutaties zijn, met een zeer variabele fenotypische expressie.4
De familie die wij hier presenteren heeft een nonsense mutatie in de regio die codeert voor het actieve domein van het eiwit, resulterend in de eliminatie van de laatste 14 aminozuren. Dit is de tweede nonsense mutatie die in de literatuur is beschreven.3 De eerste is een soortgelijke mutatie in het aminozuur dat onmiddellijk voorafgaat aan het gemuteerde aminozuur in de familie die wij hier beschrijven (p.Tyr487*/c.1461T>G), wat suggereert dat beide aanleiding geven tot mutante monomeren en functionele haploinsufficiëntie van GDF5, en zo BDC veroorzaken.
Financiering
Deze studie werd gedeeltelijk gefinancierd door de projecten PI13/00467 en PI13/01295, geïntegreerd in het R&D&I plan 2013-2016 van de Spaanse regering en medegefinancierd door de Deputy General-Directorate of Research Evaluation and Promotion van het Instituto de Salud Carlos III (ISCIII), het Europees Fonds voor Regionale Ontwikkeling (EFRO) en het Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición (Netwerkcentrum voor biomedisch onderzoek naar de fysiopathologie van zwaarlijvigheid en voeding), ISCIII, Madrid.