Termín brachydaktylie zahrnuje skupinu kostních dysplazií postihujících záprstní a/nebo záprstní/metatarzální kosti rukou a nohou. Rozlišuje se 5 typů (A-E) a několik podtypů (A1-A4; E1-E3). Dědičnost je autozomálně dominantní s různou penetrancí. Případy s recesivní dědičností jsou velmi vzácné.
Brachydaktylie typu C (BDC) je charakterizována zkrácením středních článků druhého, třetího a pátého prstu a prvního metakarpu a může se projevovat také ulnární deviací ukazováku, polydaktylií nebo výraznou hypersegmentací proximálních nebo středních článků druhého a třetího prstu. Čtvrtý prst je nejméně postižený a obvykle nejdelší.1-3 „Andělsky tvarované“ falangy (obr. 1A), ačkoli jsou charakteristické pro BDC, nejsou patognomonické, protože se mohou vyskytovat také u poruchy známé jako andělsky tvarovaná falangová epifyzární dysplazie. Je možné, že tato porucha a BDC jsou součástí stejného klinického spektra.1 U BDC se tento znak normalizuje po dokončení fyziologického uzávěru kostí ruky a končí jako prostá brachydaktylie.1 Dalšími anomáliemi spojenými s BDC jsou malý vzrůst s opožděným kostním věkem, Madelungova deformita, dysplazie kyčlí, talipes valgus nebo equinovarus nebo absence středních článků prstů.2,3
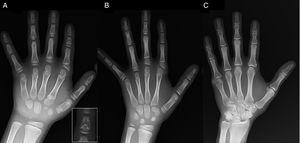
(A) Rentgenový snímek levé ruky probanda (chronologický věk 7 let; kostní věk 5 let) se zkrácením anomální první metakarpální kosti (zdvojené proximální a distální epifyzární destičky) a středních článků druhého, třetího a pátého prstu. Druhý prst vykazuje ulnární deviaci a čtvrtý prst je nejméně postižený a nejdelší na levé ruce. Proximální epifýzy druhého a třetího prstu jsou dysplastické, u druhého prstu je nápadná střední falanga ve tvaru anděla (vložka v A). (B). Rentgenový snímek levé ruky sestry probanda (ve věku 5,5 roku bez opoždění kostního věku) se zkrácenými středními falangami druhého, třetího a pátého prstu a normálním čtvrtým prstem. V tomto případě byla první záprstní kost normální. Nejvýraznějším znakem je trojúhelníkový tvar proximální epifýzy proximální falangy druhého prstu, podobný jako u bratra, a lichoběžníkový tvar falangy ve středním nártu. Stejně jako u bratra vykazuje druhý prst ulnární deviaci. (C) Rentgenový snímek levé ruky otce probanda, na kterém je patrný pouze kostěný zbytek postaxiální hexadaktylie, která byla v dětství chirurgicky korigována.
Prezentujeme případ chlapce ve věku 7 let s radiologickými znaky kompatibilními s BDC odeslaného k posouzení malého vzrůstu (z-skóre, -1,8). Jeho otec měl jednostrannou postaxiální polydaktylii, která byla oboustranně přítomna i u strýce z otcovy strany. Radiografické vyšetření levé ruky a zápěstí odhalilo kostní věk, který byl o 2 roky nižší než chronologický věk, a anomálie, které si vyžádaly provedení skeletálního vyšetření. Vyšetření odhalilo anomálie na rukou (obr. 1A) a jemné abnormality na nohou (mírná epifyzární dysplazie v proximálních článcích některých prstů). Rentgenologické vyšetření sestry ve věku 6 let odhalilo na rukou podobné léze jako u probanda, i když méně výrazné (obr. 1B). Rentgenový snímek ruky otce (obr. 1C) odhalil pouze kostní pozůstatek postaxiální hexadaktylie, která byla v dětství chirurgicky korigována.
Sekvenování genu GDF5 u probanda odhalilo heterozygotní bodovou substituci v exonu 2 (c.1462A>T), jejímž výsledkem je předčasný stop kodon (p.Lys488*, nonsense mutace) a zkrácený protein o 14 aminokyselin kratší než divoký protein (obr. 2A). Tato mutace byla nalezena také u otce a sestry pacienta, ale ne u zdravé matky (obr. 2B). Obr. 2C ukazuje rodokmen rodiny. Jedná se o novou variantu, která je pravděpodobně patogenní a má autozomálně dominantní vzorec dědičnosti.
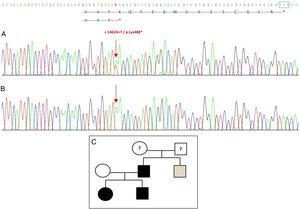
Zjištěná nová mutace v genu GDF5. (A) Sekvence genu exonu 2 u probanda, ukazující mutaci c.1462A>T, která vede k předčasnému stop kodonu a zkrácenému proteinu (p.Lys488*) (normální polohu stop kodonu jsme označili zeleným čtvercem). (B) Normální sekvence stejné oblasti u zdravé matky. (C) Rodokmen rodiny: otec a obě děti (černě) mají potvrzenou mutaci v GDF5. Zdravá matka je zobrazena bíle. Šedý čtverec představuje strýce z otcovy strany s oboustrannou postaxiální polydaktylií, který má pravděpodobně mutaci.
Růstový diferenciační faktor 5 (GDF5) je úzce spojen s kostními morfogenetickými proteiny a patří do nadrodiny transformujícího růstového faktoru β, přičemž se podílí na embryonálním vývoji kostry a kloubů.4 Gen GDF5 je mutačním ohniskem pro poruchy spojené s malformacemi skeletu.5 Většina homozygotních nebo složených heterozygotních mutací je spojena se závažnými onemocněními: Chondrodysplazie typu Grebe (OMIM 200700), akromesomelická dysplazie typu Hunter-Thomson (OMIM 201250) nebo Du Panův syndrom (OMIM 228900). Na druhé straně heterozygotní mutace spojené s mírnějšími skeletálními dysplaziemi: proximální symfalangismus 1 B (OMIM 615298) a syndrom mnohočetné synostózy typu 2 (OMIM 610017), oba spojené s missense mutacemi se ziskem funkce, a brachydaktylie typu A1 a A2, rovněž spojené s missense mutacemi, ale se ztrátou funkce.5 Brachydaktylie typu C je spojena s heterozygotními mutacemi se ztrátou funkce, ačkoli byly zaznamenány i 3 případy s recesivní dědičností.6 Většina mutací spojených s BDC jsou frameshift mutace v prodoméně části genu, zatímco většina mutací ve zralé doméně jsou missense mutace s velmi variabilním fenotypovým projevem.4
Rodina, kterou zde představujeme, má nonsense mutaci v oblasti, která kóduje aktivní doménu proteinu, což vede k odstranění jeho posledních 14 aminokyselin. Jedná se o druhou nonsensovou mutaci postihující aktivní zralou doménu popsanou v literatuře.3 První je podobná mutace v aminokyselině bezprostředně předcházející té mutované v rodině, kterou zde popisujeme (p.Tyr487*/c.1461T>G), což naznačuje, že obě dávají vzniknout mutantním monomerům a funkční haploinsuficienci GDF5, a způsobují tak BDC.
Financování
Tato studie byla částečně financována z projektů PI13/00467 a PI13/01295, začleněných do plánu R&D&I španělské vlády na období 2013-2016 a spolufinancovaných náměstkem generálního ředitelství pro hodnocení a podporu výzkumu Instituto de Salud Carlos III (ISCIII), Evropského fondu pro regionální rozvoj (ERDF) a Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición (Síťové centrum biomedicínského výzkumu fyziopatologie obezity a výživy ), ISCIII, Madrid.