O termo braquidactilia abrange um grupo de displasias ósseas envolvendo as falanges e/ou metacarpianos/ ossos dos pés e mãos. Existem 5 tipos (A-E) e vários subtipos (A1-A4; E1-E3). Possui um padrão autossômico dominante de herança com penetração variável. Casos com herança recessiva são extremamente raros.
Braquidactilia tipo C (BDC) é caracterizada por um encurtamento das falanges médias do segundo, terceiro e quinto dedos e do primeiro metacarpo, podendo também apresentar desvio ulnar do dedo indicador, polidactilia, ou uma hipersegmentação distinta das falanges proximais ou médias do segundo e terceiro dedos. O quarto dígito é o menos afetado e geralmente o mais longo.1-3 As falanges “em forma de anjo” (Fig. 1A), embora características do BDC, não são patognomônicas, pois também podem ocorrer no distúrbio conhecido como displasia falango-epifisária em forma de anjo. É possível que esta desordem e o BDC façam parte do mesmo espectro clínico.1 No BDC, esta característica se normaliza quando o fechamento dos ossos da mão é completado, terminando como braquidactilia simples.1 Outras anomalias associadas ao BDC são: baixa estatura com retardo na idade óssea, deformidade de Madelung, displasia de quadril, talipes valgo ou eqüinovaro ou ausência de falanges médias nos dedos dos pés.2,3
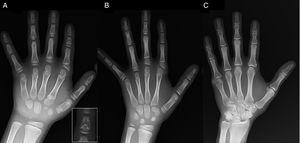
(A) Radiografia da mão esquerda da sonda (idade cronológica, 7 anos; idade óssea, 5 anos), com encurtamento de um primeiro metacarpo anômalo (dupla placa epifisária proximal e distal) e das falanges médias do segundo, terceiro e quinto dedos. O segundo dedo apresenta desvio ulnar, sendo o quarto dedo o menos afectado e o mais longo da mão esquerda. As epífises proximais do segundo e terceiro dedos são displásicas, com uma falange média em forma de anjo visível no segundo dedo (inserção em A). (B). Radiografia da mão esquerda da irmã da sonda (com idade de 5½ anos, sem atraso na idade óssea), com encurtamento das falanges médias do segundo, terceiro e quinto dedos e um quarto dedo normal. Neste caso, o primeiro metacarpo era normal. A característica mais marcante é a forma triangular da epífise proximal da falange proximal do segundo dedo, semelhante à do irmão, e a forma trapezoidal da falange no achador médio. Tal como o irmão, o segundo dedo apresenta um desvio ulnar. (C) Radiografia da mão esquerda do pai da sonda, que revela apenas um remanescente ósseo de uma hexadactilia pós-axial que foi corrigido cirurgicamente na infância.
Apresentamos o caso de um menino de 7 anos com características radiológicas compatíveis com o BDC referido para avaliação de baixa estatura (z-score, -1,8). Seu pai tinha polidactilia unilateral pós-axial, que também estava presente bilateralmente em um tio paterno. O exame radiográfico da mão e punho esquerdos revelou uma idade óssea 2 anos mais nova do que a idade cronológica e anomalias que motivaram a realização de um levantamento esquelético. O exame detectou anomalias nas mãos (Fig. 1A) e alterações sutis nos pés (displasia epifisária leve nas falanges proximais de alguns dedos dos pés). A avaliação radiológica de uma irmã de 6 anos de idade revelou lesões na mão semelhantes às da sonda, embora menos pronunciadas (Fig. 1B). Uma radiografia da mão do pai (Fig. 1C) revelou apenas um remanescente ósseo da hexadactilia pós-axial que havia sido corrigido cirurgicamente na infância.
Sequenciamento do gene GDF5 na probanda revelou uma substituição do ponto heterozigoto no exon 2 (c.1462A>T) resultando em um códão de parada prematura (p.Lys488*, mutação sem sentido) e uma proteína truncada 14 aminoácidos mais curta que a proteína selvagem (Fig. 2A). Esta mutação também foi encontrada no pai e na irmã do paciente, mas não na mãe saudável (Fig. 2B). Fig. 2C mostra o pedigree da família. Esta é uma variante nova que provavelmente é patogênica e com um padrão autossômico dominante de herança.
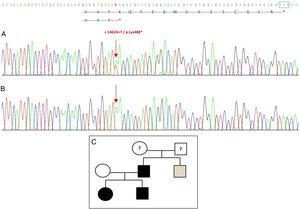
Nova mutação detectada no gene GDF5. (A) Seqüência do gene do exon 2 na probanda, mostrando uma mutação c.1462A>T que resulta em um códão de parada prematura e uma proteína truncada (p.Lys488*) (indicamos a posição normal do códão de parada com um quadrado verde). (B) Sequência normal da mesma região na mãe saudável. (C) Pedigree da família: o pai e ambos os filhos, em preto, têm uma mutação confirmada em GDF5. A mãe saudável aparece em branco. O quadrado cinza representa um tio paterno com polidactilia bilateral pós-axial que provavelmente tem a mutação.
Fator de diferenciação de crescimento 5 (GDF5) está intimamente associado a proteínas morfogenéticas ósseas e pertence ao fator de crescimento transformador β superfamília, com envolvimento no desenvolvimento do esqueleto embrionário e das articulações.4 O gene GDF5 é um hotspot mutacional para doenças associadas a malformações esqueléticas.5 A maioria das mutações homozigotos ou heterozigotos compostos estão associados a doenças graves: Condrodisplasia tipo Grebe (OMIM 200700), displasia acromesomélica tipo Hunter-Thomson (OMIM 201250) ou síndrome de Du Pan (OMIM 228900). Por outro lado, mutações heterozigotas associadas a displasias esqueléticas mais brandas: simfalangismo proximal 1 B (OMIM 615298) e síndrome de sinostose múltipla tipo 2 (OMIM 610017), ambas associadas a mutações de missense com ganho de função, e braquidactilia tipo A1 e A2, também associadas a mutações de missense, mas com perda de função.5 A braquidactilia tipo C está associada a mutações heterozigotas com perda de função, embora 3 casos com herança recessiva também tenham sido relatados.6 A maioria das mutações associadas ao BDC são mutações frameshift na parte prodomínio do gene, enquanto a maioria das mutações no domínio maduro são mutações missense, com uma expressão fenotípica altamente variável.4
A família que apresentamos aqui tem uma mutação sem sentido na região que codifica o domínio ativo da proteína, resultando na eliminação de seus últimos 14 aminoácidos. Esta é a segunda mutação sem sentido que afeta o domínio maduro ativo descrito na literatura.3 A primeira é uma mutação semelhante no aminoácido imediatamente anterior à mutação na família que descrevemos aqui (p.Tyr487*/c.1461T>G), o que sugere que ambos dão origem a monómeros mutantes e haploinsuficiência funcional do GDF5, causando assim o BDC.
Financiamento
Este estudo foi parcialmente financiado pelos projetos PI13/00467 e PI13/01295, integrados no Plano 2013-2016 R&D&I do Governo Espanhol e co-financiado pelo Direção Geral Adjunta de Avaliação e Promoção da Pesquisa do Instituto de Salud Carlos III (ISCIII), o Fundo Europeu de Desenvolvimento Regional (FEDER) e o Centro de Investigação Biomédica em Rede sobre Fisiopatologia da Obesidade e da Nutrição, ISCIII, Madrid.