Le terme brachydactylie englobe un groupe de dysplasies osseuses impliquant les phalanges et/ou les os métacarpiens/métatarsiens des mains et des pieds. Il existe 5 types (A-E) et plusieurs sous-types (A1-A4 ; E1-E3). Le mode de transmission est autosomique dominant avec une pénétrance variable. Les cas d’hérédité récessive sont extrêmement rares.
La brachydactylie de type C (BDC) se caractérise par un raccourcissement des phalanges moyennes des deuxième, troisième et cinquième doigts et du premier métacarpien, et peut également se présenter avec une déviation ulnaire de l’index, une polydactylie ou une hypersegmentation distinctive des phalanges proximales ou moyennes des deuxième et troisième doigts. Le quatrième doigt est le moins affecté et généralement le plus long.1-3 Les phalanges en forme d’ange (Fig. 1A), bien que caractéristiques du BDC, ne sont pas pathognomoniques, car elles peuvent également se produire dans le trouble connu sous le nom de dysplasie phalango-épiphysaire en forme d’ange. Il est possible que ce trouble et le BDC fassent partie du même spectre clinique.1 Dans le cas du BDC, cette caractéristique se normalise lorsque la fermeture physaire des os de la main est achevée, se terminant par une brachydactylie simple.1 Les autres anomalies associées au BDC sont une petite taille avec un âge osseux retardé, une déformation de Madelung, une dysplasie de la hanche, un talipes valgus ou équinovarus ou l’absence de phalanges moyennes dans les orteils2,3.
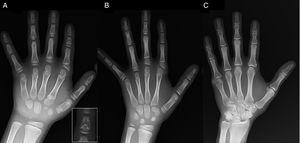
(A) Radiographie de la main gauche du proband (âge chronologique, 7 ans ; âge osseux, 5 ans), avec raccourcissement d’un premier métacarpien anormal (double plaque épiphysaire proximale et distale) et des phalanges moyennes des deuxième, troisième et cinquième doigts. Le deuxième doigt présente une déviation ulnaire, et le quatrième doigt est le moins affecté et le plus long de la main gauche. Les épiphyses proximales du deuxième et du troisième doigt sont dysplasiques, avec une phalange moyenne en forme d’ange bien visible dans le deuxième doigt (encart en A). (B). Radiographie de la main gauche de la sœur du proband (âgée de 5½ ans sans retard d’âge osseux), avec un raccourcissement des phalanges moyennes des deuxième, troisième et cinquième doigts et un quatrième doigt normal. Dans ce cas, le premier métacarpien était normal. Le trait le plus marquant est la forme triangulaire de l’épiphyse proximale de la phalange proximale du deuxième doigt, semblable à celle du frère, et la forme trapézoïdale de la phalange du milieu. Comme son frère, son deuxième doigt présente une déviation ulnaire. (C) Radiographie de la main gauche du père du proband, qui ne révèle qu’un vestige osseux d’une hexadactylie post-axiale corrigée chirurgicalement dans l’enfance.
Nous présentons le cas d’un garçon âgé de 7 ans avec des caractéristiques radiologiques compatibles avec le BDC référé pour une évaluation de la petite taille (z-score, -1,8). Son père avait une polydactylie post-axiale unilatérale, qui était également présente de manière bilatérale chez un oncle paternel. L’examen radiographique de la main et du poignet gauches a révélé un âge osseux inférieur de 2 ans à l’âge chronologique et des anomalies qui ont motivé la réalisation d’une étude du squelette. L’étude a détecté des anomalies dans les mains (Fig. 1A) et de subtiles anomalies dans les pieds (légère dysplasie épiphysaire dans les phalanges proximales de certains orteils). L’évaluation radiologique d’une sœur âgée de 6 ans a révélé des lésions de la main similaires à celles du proband, bien que moins prononcées (Fig. 1B). Une radiographie de la main du père (Fig. 1C) n’a révélé qu’un vestige osseux de l’hexadactylie post-axiale qui avait été corrigée chirurgicalement dans l’enfance.
Le séquençage du gène GDF5 chez le proband a révélé une substitution ponctuelle hétérozygote dans l’exon 2 (c.1462A>T) entraînant un codon stop prématuré (p.Lys488*, mutation non-sens) et une protéine tronquée de 14 acides aminés plus courte que la protéine sauvage (Fig. 2A). Cette mutation a également été trouvée chez le père et la sœur du patient, mais pas chez la mère saine (Fig. 2B). La figure 2C montre le pedigree de la famille. Il s’agit d’une nouvelle variante probablement pathogène avec un mode de transmission autosomique dominant.
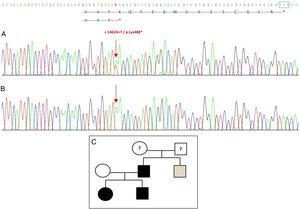
Nouvelle mutation détectée dans le gène GDF5. (A) Séquence génétique de l’exon 2 chez le proband, montrant une mutation c.1462A>T qui entraîne un codon stop prématuré et une protéine tronquée (p.Lys488*) (nous avons indiqué la position normale du codon stop par un carré vert). (B) Séquence normale de la même région chez la mère saine. (C) Pedigree de la famille : le père et les deux enfants, en noir, ont une mutation confirmée dans GDF5. La mère saine apparaît en blanc. Le carré gris représente un oncle paternel atteint de polydactylie post-axiale bilatérale et probablement porteur de la mutation.
Le facteur de différenciation de croissance 5 (GDF5) est étroitement associé aux protéines morphogénétiques osseuses et appartient à la superfamille des facteurs de croissance transformants β, avec une implication dans le développement embryonnaire du squelette et des articulations4. Le gène GDF5 est un point chaud de mutation pour les troubles associés aux malformations du squelette.5 La plupart des mutations homozygotes ou hétérozygotes composées sont associées à des maladies graves : La plupart des mutations homozygotes ou hétérozygotes composées sont associées à des maladies graves : chondrodysplasie de type Grebe (OMIM 200700), dysplasie acromésomélique de type Hunter-Thomson (OMIM 201250) ou syndrome de Du Pan (OMIM 228900). D’autre part, des mutations hétérozygotes sont associées à des dysplasies squelettiques plus légères : le symphalangisme proximal 1 B (OMIM 615298) et le syndrome de synostose multiple de type 2 (OMIM 610017), tous deux associés à des mutations faux sens avec gain de fonction, et la brachydactylie de type A1 et A2, également associée à des mutations faux sens, mais avec perte de fonction5. La brachydactylie de type C est associée à des mutations hétérozygotes avec perte de fonction, bien que trois cas d’hérédité récessive aient également été signalés6. La plupart des mutations associées au BDC sont des mutations de type frameshift dans la partie prodomaine du gène, tandis que la plupart des mutations dans le domaine mature sont des mutations missens, avec une expression phénotypique très variable.4
La famille que nous présentons ici présente une mutation non-sens dans la région qui code pour le domaine actif de la protéine, entraînant l’élimination de ses 14 derniers acides aminés. Il s’agit de la deuxième mutation non-sens affectant le domaine actif mature décrite dans la littérature.3 La première est une mutation similaire dans l’acide aminé précédant immédiatement celui muté dans la famille que nous décrivons ici (p.Tyr487*/c.1461T>G), ce qui suggère que les deux donnent lieu à des monomères mutants et à une haplo-insuffisance fonctionnelle du GDF5, causant ainsi le BDC.
Financement
Cette étude a été partiellement financée par les projets PI13/00467 et PI13/01295, intégrés dans le plan R&D&I 2013-2016 du gouvernement espagnol et cofinancés par la Direction générale adjointe d’évaluation et de promotion de la recherche de l’Instituto de Salud Carlos III (ISCIII), le Fonds européen de développement régional (FEDER) et le Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición (Centre de recherche biomédicale en réseau sur la physiopathologie de l’obésité et de la nutrition ), ISCIII, Madrid.