Tegnet brachydactyly omfatter en gruppe af knogledysplasier, der involverer phalanges og/eller metacarpal/metatarsalknogler i hænder og fødder. Der findes 5 typer (A-E) og flere undertyper (A1-A4; E1-E3). Den har et autosomalt dominerende arvelighedsmønster med variabel penetrans. Tilfælde med recessiv arv er ekstremt sjældne.
Brachydaktyli type C (BDC) er karakteriseret ved en forkortelse af de midterste phalanges i anden, tredje og femte finger og den første metacarpal, og kan også vise sig med ulnær afvigelse af pegefingeren, polydaktyli eller en udpræget hypersegmentering af de proximale eller midterste phalanges i anden og tredje finger. Det fjerde finger er mindst påvirket og normalt længst.1-3 “Engleformede” phalanges (Fig. 1A) er karakteristiske for BDC, men er ikke patognomoniske, da de også kan forekomme i den lidelse, der er kendt som angel-shaped phalango-epiphyseal dysplasi. Det er muligt, at denne lidelse og BDC er en del af det samme kliniske spektrum.1 I BDC normaliseres dette træk, når fyseal lukning af håndknoglerne er afsluttet, og det ender som simpel brachydaktyli.1 Andre anomalier forbundet med BDC er kort statur med forsinket knoglealder, Madelung-deformitet, hoftedysplasi, talipes valgus eller equinovarus eller fravær af midterste phalanges i tæerne.2,3
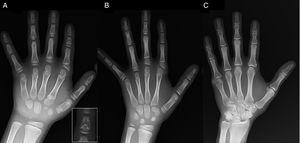
(A) Røntgenbillede af probandens venstre hånd (kronologisk alder, 7 år; knoglealder, 5 år) med forkortelse af en anomal første metacarpal (dobbelte proximale og distale epifyseplader) og de midterste phalanges af anden, tredje og femte finger. Andenfingeren udviser ulnardeviation, og fjerdefingeren er den mindst påvirkede og længst i venstre hånd. De proximale epifyser i anden og tredje finger er dysplastiske, med en iøjnefaldende engleformet midterste phalanx i anden finger (indsat i A). (B). Røntgenbillede af venstre hånd hos probandens søster (5½ år gammel uden forsinkelse i knoglealderen) med forkortelse af de midterste phalanges i anden, tredje og femte finger og en normal fjerde finger. I dette tilfælde var den første mellemfinger normal. Det mest iøjnefaldende træk er den trekantede form af den proximale epifyse på andenfingerens proximale phalanx, der ligner broderens, og den trapezformede form af phalangen i den midterste finder. Ligesom hendes bror udviser hendes andenfinger ulnær deviation. (C) Røntgenbillede af venstre hånd hos probandens far, som kun afslører en knoklet rest af en postaxial hexadaktyli, der blev kirurgisk korrigeret i barndommen.
Vi præsenterer et tilfælde af en dreng på 7 år med radiologiske træk forenelige med BDC henvist til evaluering af kort statur (z-score, -1,8). Hans far havde unilateral postaksial polydaktyli, som også var til stede bilateralt hos en faderlig onkel. Radiografisk undersøgelse af venstre hånd og håndled afslørede en knoglealder, der var 2 år yngre end den kronologiske alder, og anomalier, som gav anledning til at foretage en skeletundersøgelse. Undersøgelsen afslørede anomalier i hænderne (Fig. 1A) og subtile abnormiteter i fødderne (mild epifyseal dysplasi i de proximale phalanges på nogle tæer). Den radiologiske vurdering af en søster på 6 år afslørede læsioner i hånden, der lignede dem hos probandens, selv om de var mindre udtalte (Fig. 1B). Et røntgenbillede af faderens hånd (Fig. 1C) afslørede kun en knoglerest af den postaxiale hexadaktyli, der var blevet kirurgisk korrigeret i barndommen.
Sekventering af GDF5-genet hos probandens viste en heterozygot punktsubstitution i exon 2 (c.1462A>T), hvilket resulterede i et for tidligt stopkodon (p.Lys488*, nonsense-mutation) og et afkortet protein, der er 14 aminosyrer kortere end det vilde protein (Fig. 2A). Denne mutation blev også fundet hos patientens far og søster, men ikke hos den raske mor (Fig. 2B). Fig. 2C viser familiens stamtavle. Der er tale om en ny variant, der sandsynligvis er patogen, og med et autosomalt dominerende arvelighedsmønster.
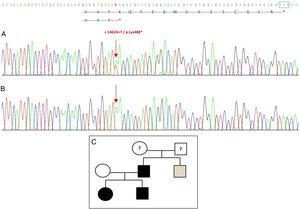
Ny mutation påvist i GDF5-genet. (A) Gensekvens af exon 2 hos proband, der viser en c.1462A>T-mutation, som resulterer i et for tidligt stopkodon og et afkortet protein (p.Lys488*) (vi har angivet den normale placering af stopkodonet med en grøn firkant). (B) Normal sekvens af den samme region hos den raske mor. (C) Stamtavle for familien: Faderen og begge børn, i sort, har en bekræftet mutation i GDF5. Den raske mor er vist i hvidt. Den grå firkant repræsenterer en onkel på faderens side med bilateral postaksial polydaktyli, som sandsynligvis har mutationen.
Vækstdifferentieringsfaktor 5 (GDF5) er tæt forbundet med knoglemorfogenetiske proteiner og hører til den transformerende vækstfaktor β-superfamilie, med er involveret i embryonal skelet- og ledudvikling.4 GDF5-genet er et mutationelt hotspot for sygdomme, der er forbundet med skeletfejldannelser.5 De fleste homozygote eller sammensatte heterozygote mutationer er forbundet med alvorlige sygdomme: De hyppigst heterotomierede eller heterotomierede mutationer er forbundet med alvorlige sygdomme som chondrodysplasi af Grebe-typen (OMIM 200700), akromesomelisk dysplasi af Hunter-Thomson-typen (OMIM 201250) eller Du Pan-syndromet (OMIM 228900). På den anden side er heterozygote mutationer forbundet med mildere skeletdysplasier: proximal symphalangisme 1 B (OMIM 615298) og multipel synostose syndrom type 2 (OMIM 610017), begge forbundet med missense-mutationer med funktionsforøgelse, og brachydactyly type A1 og A2, også forbundet med missense-mutationer, men med funktionstab5 . Brachydactyly type C er associeret med heterozygote mutationer med tab af funktion, selv om der også er rapporteret 3 tilfælde med recessiv arv.6 De fleste mutationer, der er forbundet med BDC, er frameshift-mutationer i prodomænedelen af genet, mens de fleste mutationer i det modne domæne er missense-mutationer, med et meget variabelt fænotypisk udtryk.4
Den familie, som vi præsenterer her, har en nonsens-mutation i den region, der koder for proteinets aktive domæne, hvilket resulterer i eliminering af dets sidste 14 aminosyrer. Dette er den anden nonsense-mutation, der påvirker det aktive modne domæne, som er beskrevet i litteraturen.3 Den første er en lignende mutation i den aminosyre, der går umiddelbart forud for den, der er muteret i den familie, som vi beskriver her (p.Tyr487*/c.1461T>G), hvilket tyder på, at begge giver anledning til muterede monomerer og funktionel haploinsufficiens af GDF5 og dermed forårsager BDC.
Finansiering
Denne undersøgelse blev delvist finansieret af projekterne PI13/00467 og PI13/01295, der er integreret i den spanske regerings R&D&I-plan 2013-2016 og samfinansieret af Deputy General-Directorate of Research Evaluation and Promotion of the Instituto de Salud Carlos III (ISCIII), Den Europæiske Fond for Regionaludvikling (EFRU) og Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición (Center for biomedicinsk forskning i netværk om fysiopatologi i forbindelse med fedme og ernæring), ISCIII, Madrid.