A brachydactylia kifejezés a kezek és lábak ujjperceit és/vagy kézközépcsontjait érintő csontdiszpláziák egy csoportját foglalja magában. Öt típusa (A-E) és több altípusa (A1-A4; E1-E3) különböztethető meg. Autoszomális domináns öröklődési mintázatú, változó penetrációval. A recesszív öröklődésű esetek rendkívül ritkák.
A C típusú brachydactylia (BDC) a második, harmadik és ötödik ujj középső ujjpercének és az első kézközépcsontnak a megrövidülésével jellemezhető, és a mutatóujj ulnaris eltérése, polydactylia, vagy a második és harmadik ujj proximális vagy középső ujjpercének jellegzetes hipersegmentációja is előfordulhat. A negyedik ujjperc a legkevésbé érintett és általában a leghosszabb.1-3 Az “angyal alakú” ujjpercek (1A. ábra), bár a BDC-re jellemzőek, nem patognomikusak, mivel az angyal alakú phalango-epifízis diszplázia néven ismert rendellenességben is előfordulhatnak. Lehetséges, hogy ez a rendellenesség és a BDC ugyanannak a klinikai spektrumnak a részét képezi.1 A BDC esetében ez a jellegzetesség normalizálódik, amikor a kézcsontok fízisének záródása befejeződik, és egyszerű brachydaktíliaként végződik.1 A BDC-vel kapcsolatos egyéb rendellenességek a késleltetett csontkorral járó alacsony termet, a Madelung-deformitás, a csípődiszplázia, a talipes valgus vagy equinovarus vagy a lábujjak középső ujjperceinek hiánya.2,3,3.
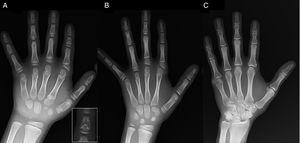
(A) Röntgenfelvétel a probandus bal kezéről (kronológiai életkor 7 év; csontkor 5 év), az anomális első metacarpalis (kettős proximalis és distalis epifízislemez) és a második, harmadik és ötödik ujj középső ujjpercének rövidülésével. A második ujj ulnaris eltérést mutat, és a negyedik ujj a legkevésbé érintett és a bal kézben a leghosszabb. A második és a harmadik ujj proximális epifízisei diszpláziásak, a második ujjban feltűnő angyal alakú középső ujjpercekkel (A betét). (B). Röntgenfelvétel a probandus testvér bal kezéről (5 és fél éves, csontkésés nélkül), a második, harmadik és ötödik ujj középső ujjpercének megrövidülésével és normális negyedik ujjal. Ebben az esetben az első kézközépcsont normális volt. A legkiemelkedőbb jellemző a második ujj proximalis epifízisének háromszög alakja, amely hasonló a testvéréhez, és a középső ujjperccsont trapéz alakja. A bátyjához hasonlóan a második ujj is ulnaris deviációt mutat. (C) A probandus apjának bal kezéről készült röntgenfelvétel, amelyen csak a gyermekkorban műtétileg korrigált posztaxiális hexadactylia csontos maradványa látható.
Egy 7 éves fiú esetét mutatjuk be, akinek radiológiai jellemzői összeegyeztethetők a BDC-vel, és akit rövid termet (z-score, -1,8) értékelése céljából utaltak be. Apjának egyoldali postaxiális polydactyliája volt, amely apai nagybátyjánál szintén kétoldali polydactylia volt. A bal kéz és a csukló röntgenvizsgálata a kronológiai életkornál 2 évvel fiatalabb csontkort és olyan rendellenességeket mutatott ki, amelyek csontvázvizsgálat elvégzésére késztettek. A vizsgálat a kezekben rendellenességeket (1A. ábra) és a lábakban finom rendellenességeket (enyhe epifízisdiszplázia néhány lábujj proximális ujjpercében) mutatott ki. Egy 6 éves testvér radiológiai vizsgálata a kezeken a próbanduséhoz hasonló, bár kevésbé kifejezett elváltozásokat mutatott ki (1B. ábra). Az apa kezének röntgenfelvétele (1C. ábra) csak a gyermekkorban műtétileg korrigált poszt-axiális hexadaktíliának egy csontmaradványát mutatta ki.
A GDF5 gén szekvenálása a probandusnál heterozigóta pontszerű szubsztitúciót mutatott ki a 2. exonban (c.1462A>T), amely egy korai stop kodont (p.Lys488*, nonszensz mutáció) és a vad fehérjénél 14 aminosavval rövidebb csonka fehérjét eredményezett (2A ábra). Ez a mutáció a beteg apjában és nővérében is megtalálható volt, de az egészséges anyában nem (2B ábra). A 2C. ábra a család pedigréjét mutatja. Ez egy új variáns, amely valószínűleg patogén és autoszomális domináns öröklődési mintázatú.
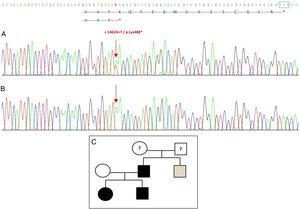
A GDF5 génben kimutatott új mutáció. (A) A probandus 2. exonjának génszekvenciája, amely a c.1462A>T mutációt mutatja, amely egy korai stop kodont és egy csonka fehérjét (p.Lys488*) eredményez (a stop kodon normál helyzetét zöld négyzettel jelöltük). (B) Ugyanezen régió normális szekvenciája az egészséges anyában. (C) A család törzskönyve: az apa és mindkét gyermek (fekete színnel) megerősített mutációval rendelkezik a GDF5-ben. Az egészséges anya fehérrel szerepel. A szürke négyzet egy apai nagybácsit ábrázol, akinek kétoldali poszt-axiális polydaktíliája van, és aki valószínűleg rendelkezik a mutációval.
A növekedési differenciálódási faktor 5 (GDF5) szorosan kapcsolódik a csontmorfogenetikai fehérjékhez és a transzformáló növekedési faktor β szupercsaládhoz tartozik, az embrionális csontváz és az ízületek fejlődésében vesz részt.4 A GDF5 gén a csontrendszeri rendellenességekkel járó rendellenességek mutációs gócpontja.5 A legtöbb homozigóta vagy összetett heterozigóta mutáció súlyos betegségekkel jár: Grebe-típusú chondrodysplasia (OMIM 200700), Hunter-Thomson-típusú akromesomelikus diszplázia (OMIM 201250) vagy Du Pan-szindróma (OMIM 228900). Másrészt a heterozigóta mutációk enyhébb csontrendszeri diszpláziákhoz társulnak: proximális szimphalangizmus 1 B (OMIM 615298) és 2-es típusú multiplex szinosztózis szindróma (OMIM 610017), mindkettő miszense mutációval és funkcióvesztéssel társul, valamint brachydactylia A1 és A2 típusú, szintén miszense mutációkkal, de funkcióvesztéssel.5. 5 A C típusú brachydactylia funkcióvesztéssel járó heterozigóta mutációkhoz társul, bár 3 recesszív öröklődésű esetről is beszámoltak6. A BDC-vel társuló mutációk többsége frameshift mutáció a gén prodomain részében, míg az érett doménben a legtöbb mutáció missense mutáció, igen változó fenotípusos kifejeződéssel.4
Az általunk bemutatott családban nonsense mutáció található a fehérje aktív doménjét kódoló régióban, ami a fehérje utolsó 14 aminosavának eltűnését eredményezi. Ez a második, az aktív érett domént érintő nonszensz mutáció, amelyet az irodalomban leírtak.3 Az első egy hasonló mutáció az általunk ismertetett családban mutálódott aminosavat közvetlenül megelőző aminosavban (p.Tyr487*/c.1461T>G), ami arra utal, hogy mindkettő mutáns monomereket és a GDF5 funkcionális haploinsufficienciáját eredményezi, és így BDC-t okoz.
Finanszírozás
Ezt a vizsgálatot részben a PI13/00467 és PI13/01295 projektek finanszírozták, amelyek a spanyol kormány 2013-2016-os R&D&I tervébe integrálódtak, és amelyeket az Instituto de Salud Carlos III (ISCIII) Kutatásértékelési és Kutatásösztönzési Főigazgatóságának helyettese társfinanszírozott, az Európai Regionális Fejlesztési Alap (ERFA) és a Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición (Biomedical Research Networking Centre on the Physiopathology of Obesity and Nutrition ), ISCIII, Madrid.