Termin brachydactyly obejmuje grupę dysplazji kostnych obejmujących paliczki i/lub kości śródręcza/śródstopia rąk i stóp. Wyróżnia się 5 typów (A-E) i kilka podtypów (A1-A4; E1-E3). Charakteryzuje się dziedziczeniem autosomalnym dominującym o zmiennej penetracji. Przypadki dziedziczone recesywnie są niezwykle rzadkie.
Brachydaktylia typu C (BDC) charakteryzuje się skróceniem środkowych paliczków drugiego, trzeciego i piątego palca oraz pierwszego śródręcza, może również występować z odchyleniem łokciowym palca wskazującego, polidaktylią lub charakterystyczną hipersegmentacją proksymalnych lub środkowych paliczków drugiego i trzeciego palca. Czwarty palec jest najmniej dotknięty chorobą i zazwyczaj najdłuższy.1-3 Paliczki w kształcie anioła (ryc. 1A), choć charakterystyczne dla BDC, nie są patognomoniczne, ponieważ mogą występować również w zaburzeniu znanym jako dysplazja anielsko-epifizjologiczna paliczków. Jest możliwe, że to zaburzenie i BDC są częścią tego samego spektrum klinicznego.1 W BDC cecha ta normalizuje się po zakończeniu procesu zamykania kostnego kości dłoni, co kończy się brachydycezją prostą.1 Inne anomalie związane z BDC to niski wzrost z opóźnionym wiekiem kostnym, deformacja Madelunga, dysplazja stawów biodrowych, talipes valgus lub equinovarus lub brak paliczków środkowych w palcach stóp.2,3
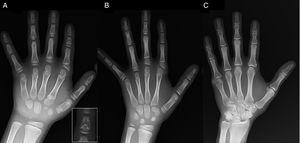
Przedstawiamy przypadek chłopca w wieku 7 lat z radiologicznymi cechami zgodnymi z BDC, skierowanego do oceny krótkiego wzrostu (z-score, -1,8). U ojca chłopca stwierdzono jednostronną polidaktylię poosiową, która występowała również obustronnie u wujka ze strony ojca. Badanie radiograficzne lewej ręki i nadgarstka wykazało wiek kostny o 2 lata niższy od wieku chronologicznego oraz anomalie, które skłoniły do wykonania badania szkieletu. W badaniu tym wykryto anomalie w obrębie rąk (ryc. 1A) oraz subtelne nieprawidłowości w obrębie stóp (łagodna dysplazja nasadowa w paliczkach bliższych niektórych palców). Ocena radiologiczna siostry w wieku 6 lat wykazała zmiany w obrębie dłoni podobne do występujących u probanta, choć mniej nasilone (ryc. 1B). Zdjęcie radiologiczne ręki ojca (ryc. 1C) ujawniło jedynie pozostałość kostną po korygowanej chirurgicznie w dzieciństwie heksadaktylii poosiowej.
Sekwencjonowanie genu GDF5 u probanta ujawniło heterozygotyczną substytucję punktową w eksonie 2 (c.1462A>T) skutkującą przedwczesnym kodonem stop (p.Lys488*, mutacja nonsensowna) i białkiem okrojonym o 14 aminokwasów krótszym od białka dzikiego (ryc. 2A). Mutację tę stwierdzono również u ojca i siostry pacjentki, ale nie u zdrowej matki (ryc. 2B). Ryc. 2C przedstawia rodowód rodziny. Jest to nowy wariant, prawdopodobnie patogenny, z autosomalnym dominującym wzorcem dziedziczenia.
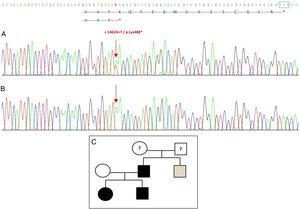
Nowa mutacja wykryta w genie GDF5. (A) Sekwencja genowa eksonu 2 u probanta, pokazująca mutację c.1462A>T, która skutkuje przedwczesnym kodonem stop i obciętym białkiem (p.Lys488*) (zaznaczyliśmy normalną pozycję kodonu stop zielonym kwadratem). (B) Prawidłowa sekwencja tego samego regionu u zdrowej matki. (C) Pedigree rodziny: ojciec i oboje dzieci, w kolorze czarnym, mają potwierdzoną mutację w GDF5. Zdrowa matka jest w kolorze białym. Szary kwadrat przedstawia ojcowskiego wujka z obustronną polidaktylią poosiową, który prawdopodobnie ma mutację.
Czynnik różnicowania wzrostu 5 (GDF5) jest ściśle związany z białkami morfogenetycznymi kości i należy do nadrodziny transformującego czynnika wzrostu β, z jest zaangażowany w embrionalny rozwój szkieletu i stawów.4. Gen GDF5 jest mutacyjnym hotspotem dla zaburzeń związanych z deformacjami szkieletu.5 Większość mutacji homozygotycznych lub złożonych heterozygotycznych wiąże się z ciężkimi chorobami: chondrodysplazją typu Grebe (OMIM 200700), dysplazją akromesomeliczną typu Hunter-Thomson (OMIM 201250) czy zespołem Du Pan (OMIM 228900). Z drugiej strony, heterozygotyczne mutacje związane z łagodniejszymi dysplazjami szkieletowymi: proksymalny symphalangizm 1 B (OMIM 615298) i zespół synostozy mnogiej typu 2 (OMIM 610017), oba związane z mutacjami typu missense z zyskiem funkcji, oraz brachydaktylia typu A1 i A2, również związane z mutacjami typu missense, ale z utratą funkcji.5 Brachydaktylia typu C jest związana z heterozygotycznymi mutacjami z utratą funkcji, chociaż opisano również 3 przypadki dziedziczenia recesywnego.6 Większość mutacji związanych z BDC to mutacje typu frameshift w części prodomeny genu, podczas gdy większość mutacji w dojrzałej domenie to mutacje typu missense, z wysoce zmienną ekspresją fenotypową.4
Rodzina, którą prezentujemy tutaj ma mutację nonsensowną w regionie, który koduje aktywną domenę białka, powodując eliminację jej ostatnich 14 aminokwasów. Jest to druga mutacja nonsensowna dotycząca aktywnej domeny dojrzałej opisana w literaturze.3 Pierwszą jest podobna mutacja w aminokwasie bezpośrednio poprzedzającym ten zmutowany w rodzinie, którą tu opisujemy (p.Tyr487*/c.1461T>G), co sugeruje, że obie dają początek zmutowanym monomerom i funkcjonalnej haploinsufficiency GDF5, powodując w ten sposób BDC.
Finansowanie
Badanie to było częściowo finansowane przez projekty PI13/00467 i PI13/01295, włączone do planu 2013-2016 R&D&I rządu hiszpańskiego i współfinansowane przez Zastępcę Dyrekcji Generalnej ds. Oceny Badań i Promocji Instituto de Salud Carlos III (ISCIII), Europejski Fundusz Rozwoju Regionalnego (ERDF) oraz Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición (Centrum Sieciowe Badań Biomedycznych nad Fizjopatologią Otyłości i Odżywiania), ISCIII, Madryt.