Termenul brahidactilie înglobează un grup de displazii osoase care implică falangele și/sau oasele metacarpiene/metatarsiene ale mâinilor și picioarelor. Există 5 tipuri (A-E) și mai multe subtipuri (A1-A4; E1-E3). Are un tipar de moștenire autosomal dominant cu penetranță variabilă. Cazurile cu moștenire recesivă sunt extrem de rare.
Brahidactilia de tip C (BDC) se caracterizează printr-o scurtare a falangelor medii ale celui de-al doilea, al treilea și al cincilea deget și ale primului metacarpian și poate prezenta, de asemenea, o deviație ulnară a degetului arătător, polidactilie sau o hipersegmentare distinctivă a falangelor proximale sau medii ale celui de-al doilea și al treilea deget. Al patrulea deget este cel mai puțin afectat și, de obicei, este cel mai lung.1-3 Falangele „în formă de înger” (Fig. 1A), deși sunt caracteristice BDC, nu sunt patognomonice, deoarece pot apărea și în tulburarea cunoscută sub numele de displazie falango-epifizară în formă de înger. Este posibil ca această tulburare și BDC să facă parte din același spectru clinic.1 În cazul BDC, această trăsătură se normalizează atunci când închiderea foselor oaselor mâinii este finalizată, încheindu-se ca o brahidactilie simplă.1 Alte anomalii asociate cu BDC sunt statura scurtă cu vârsta osoasă întârziată, deformarea Madelung, displazia șoldului, talipes valgus sau equinovarus sau absența falangelor medii la degetele de la picioare.2,3
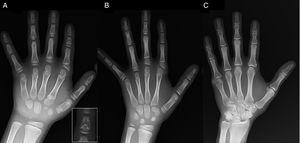
(A) Radiografie a mâinii stângi a probandului (vârsta cronologică, 7 ani; vârsta osoasă, 5 ani), cu scurtarea unui prim metacarpian anomal (plăci epifizare duble proximale și distale) și a falangelor medii ale degetelor al doilea, al treilea și al cincilea. Al doilea deget prezintă deviație ulnară, iar al patrulea deget este cel mai puțin afectat și cel mai lung la mâna stângă. Epifizele proximale ale celui de-al doilea și al treilea deget sunt displazice, cu o falangă mijlocie vizibilă în formă de înger la cel de-al doilea deget (inserție în A). (B). Radiografie a mâinii stângi a surorii probandului (în vârstă de 5½ ani, fără întârziere a vârstei osoase), cu scurtarea falangelor medii ale celui de-al doilea, al treilea și al cincilea deget și un al patrulea deget normal. În acest caz, primul metacarpian era normal. Cea mai evidentă trăsătură este forma triunghiulară a epifizei proximale a falangei proximale a celui de-al doilea deget, similară cu cea a fratelui, și forma trapezoidală a falangei din finul mijlociu. Ca și fratele ei, al doilea deget prezintă deviație ulnară. (C) Radiografie a mâinii stângi a tatălui probandului, care relevă doar o rămășiță osoasă a unei hexadactilii post-axiale care a fost corectată chirurgical în copilărie.
Prezentăm cazul unui băiat în vârstă de 7 ani cu caracteristici radiologice compatibile cu BDC, trimis pentru evaluarea unei staturi scurte (z-score, -1,8). Tatăl său avea polidactilie unilaterală post-axială, care era prezentă bilateral și la un unchi patern. Examinarea radiografică a mâinii și a încheieturii mâinii stângi a evidențiat o vârstă osoasă cu 2 ani mai mică decât vârsta cronologică și anomalii care au determinat efectuarea unui studiu scheletic. Studiul a detectat anomalii la nivelul mâinilor (Fig. 1A) și anomalii subtile la nivelul picioarelor (displazie epifizară ușoară în falangele proximale ale unor degete). Evaluarea radiologică a unei surori în vârstă de 6 ani a relevat leziuni la mână similare cu cele ale probandului, deși mai puțin pronunțate (Fig. 1B). O radiografie a mâinii tatălui (Fig. 1C) a relevat doar un rest osos al hexadactiliei post-axiale care fusese corectată chirurgical în copilărie.
Secvențierea genei GDF5 la proband a relevat o substituție punctiformă heterozigotă în exonul 2 (c.1462A>T) având ca rezultat un codon de oprire prematură (p.Lys488*, mutație fără sens) și o proteină trunchiată cu 14 aminoacizi mai scurtă decât proteina sălbatică (Fig. 2A). Această mutație a fost găsită, de asemenea, la tatăl și sora pacientului, dar nu și la mama sănătoasă (Fig. 2B). Fig. 2C prezintă pedigree-ul familiei. Aceasta este o variantă nouă, care este probabil patogenă și cu un model de moștenire autosomal dominant.
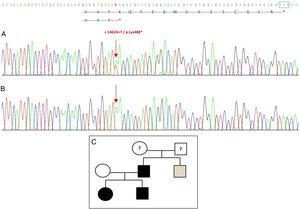
Mutație nouă detectată în gena GDF5. (A) Secvența genetică a exonului 2 la proband, care arată o mutație c.1462A>T care are ca rezultat un codon de oprire prematură și o proteină trunchiată (p.Lys488*) (am indicat poziția normală a codonului de oprire cu un pătrat verde). (B) Secvența normală a aceleiași regiuni la mama sănătoasă. (C) Pedigree al familiei: tatăl și ambii copii, în negru, au o mutație confirmată în GDF5. Mama sănătoasă apare în alb. Pătratul gri reprezintă un unchi patern cu polidactilie post-axială bilaterală, care probabil are mutația.
Factorul de diferențiere a creșterii 5 (GDF5) este strâns asociat cu proteinele morfogenetice osoase și aparține superfamiliei factorului de creștere transformant β, cu este implicat în dezvoltarea scheletului embrionar și a articulațiilor.4 Gena GDF5 este un hotspot mutațional pentru tulburările asociate cu malformații ale scheletului.5 Majoritatea mutațiilor homozigote sau heterozigote compuse sunt asociate cu boli grave: Condrodisplazia de tip Grebe (OMIM 200700), displazia acromesomelică de tip Hunter-Thomson (OMIM 201250) sau sindromul Du Pan (OMIM 228900). Pe de altă parte, mutațiile heterozigote se asociază cu displaziile scheletice mai ușoare: simfalangismul proximal 1 B (OMIM 615298) și sindromul de sinostoză multiplă tip 2 (OMIM 610017), ambele asociate cu mutații missense cu câștig de funcție, și brahidactilia tip A1 și A2, de asemenea asociate cu mutații missense, dar cu pierdere de funcție.5 Brahidacizia de tip C este asociată cu mutații heterozigote cu pierdere de funcție, deși au fost raportate și 3 cazuri cu moștenire recesivă.6 Majoritatea mutațiilor asociate cu BDC sunt mutații de tip frameshift în partea prodomeniului genei, în timp ce majoritatea mutațiilor din domeniul matur sunt mutații de tip missense, cu o expresie fenotipică foarte variabilă.4
Familia pe care o prezentăm aici are o mutație nonsens în regiunea care codifică domeniul activ al proteinei, ceea ce duce la eliminarea ultimilor 14 aminoacizi ai acesteia. Aceasta este a doua mutație fără sens care afectează domeniul activ matur descrisă în literatura de specialitate.3 Prima este o mutație similară în aminoacidul imediat anterior celui mutat în familia pe care o descriem aici (p.Tyr487*/c.1461T>G), ceea ce sugerează că ambele dau naștere la monomeri mutanți și haploinsuficiență funcțională a GDF5, provocând astfel BDC.
Finanțare
Acest studiu a fost finanțat parțial prin proiectele PI13/00467 și PI13/01295, integrate în planul R&D&I 2013-2016 al Guvernului spaniol și cofinanțat de către Direcția Generală Adjunctă de Evaluare și Promovare a Cercetării din cadrul Instituto de Salud Carlos III (ISCIII), Fondul European de Dezvoltare Regională (FEDR) și de Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición (Centrul de cercetare biomedicală în rețea privind fiziopatologia obezității și a nutriției ), ISCIII, Madrid.