Il termine brachidattilia comprende un gruppo di displasie ossee che coinvolgono le falangi e/o le ossa metacarpali/metatarsali di mani e piedi. Esistono 5 tipi (A-E) e diversi sottotipi (A1-A4; E1-E3). Ha un modello di eredità autosomica dominante con penetranza variabile. I casi con eredità recessiva sono estremamente rari.
La brachidattilia di tipo C (BDC) è caratterizzata da un accorciamento delle falangi medie del secondo, terzo e quinto dito e del primo metacarpo, e può anche presentare una deviazione ulnare del dito indice, polidattilia, o una caratteristica ipersegmentazione delle falangi prossimali o medie del secondo e terzo dito. Il quarto dito è meno colpito e di solito più lungo.1-3 Falangi “a forma di angelo” (Fig. 1A), mentre caratteristico di BDC, non sono patognomonici, in quanto possono anche verificarsi nel disordine noto come angelo a forma di falango-displasia epifisaria. È possibile che questo disturbo e la BDC facciano parte dello stesso spettro clinico.1 Nella BDC, questa caratteristica si normalizza quando la chiusura fisiologica delle ossa della mano è completata, terminando come brachidattilia semplice.1 Altre anomalie associate alla BDC sono la bassa statura con età ossea ritardata, la deformità di Madelung, la displasia dell’anca, il talipes valgus o equinovarus o l’assenza di falangi medie nelle dita dei piedi.2,3
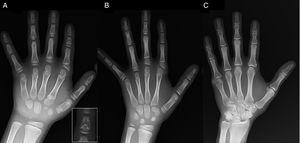
(A) Radiografia della mano sinistra del probando (età cronologica, 7 anni; età ossea, 5 anni), con accorciamento di un primo metacarpo anomalo (doppia placca epifisaria prossimale e distale) e le falangi medie del secondo, terzo e quinto dito. Il secondo dito presenta una deviazione ulnare, e il quarto dito è il meno colpito e più lungo nella mano sinistra. Le epifisi prossimali del secondo e terzo dito sono displasiche, con una vistosa falange media a forma di angelo nel secondo dito (inserto in A). (B). Radiografia della mano sinistra della sorella del probando (di 5 anni e mezzo senza ritardo nell’età ossea), con accorciamento delle falangi medie del secondo, terzo e quinto dito e un quarto dito normale. In questo caso, il primo metacarpo era normale. La caratteristica più saliente è la forma triangolare dell’epifisi prossimale della falange prossimale del secondo dito, simile a quella del fratello, e la forma trapezoidale della falange nel cercine medio. Come il fratello, il secondo dito presenta una deviazione ulnare. (C) Radiografia della mano sinistra del padre del probando, che rivela solo un residuo osseo di un esadattilia post-assiale che è stato corretto chirurgicamente nell’infanzia.
Presentiamo il caso di un bambino di 7 anni con caratteristiche radiologiche compatibili con BDC di cui per la valutazione della bassa statura (z-score, -1.8). Suo padre aveva polidattilia unilaterale post-assiale, che era anche presente bilateralmente in uno zio paterno. L’esame radiografico della mano sinistra e del polso ha rivelato un’età ossea che era 2 anni più giovane dell’età cronologica e delle anomalie che hanno richiesto l’esecuzione di un’indagine scheletrica. L’indagine ha rilevato anomalie nelle mani (Fig. 1A) e sottili anomalie nei piedi (lieve displasia epifisaria nelle falangi prossimali di alcune dita). La valutazione radiologica di una sorella di 6 anni ha rivelato lesioni nella mano simili a quelle del probando, anche se meno pronunciate (Fig. 1B). Una radiografia della mano del padre (Fig. 1C) ha rivelato solo un residuo osseo dell’esadattilia post-assiale che era stato corretto chirurgicamente nell’infanzia.
Il sequenziamento del gene GDF5 nel probando ha rivelato una sostituzione puntiforme eterozigote nell’esone 2 (c.1462A>T) con conseguente codone di stop prematuro (p.Lys488*, mutazione senza senso) e una proteina troncata di 14 aminoacidi più corta della proteina selvatica (Fig. 2A). Questa mutazione è stata trovata anche nel padre e nella sorella del paziente, ma non nella madre sana (Fig. 2B). La Fig. 2C mostra il pedigree della famiglia. Questa è una nuova variante che è probabilmente patogena e con un modello autosomico dominante di eredità.
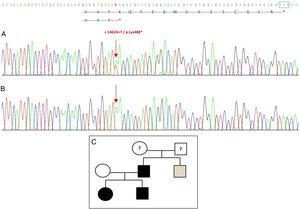
Nuova mutazione rilevata nel gene GDF5. (A) Sequenza genica dell’esone 2 nel probando, che mostra una mutazione c.1462A>T che risulta in un codone di stop prematuro e una proteina troncata (p.Lys488*) (abbiamo indicato la posizione normale del codone di stop con un quadrato verde). (B) Sequenza normale della stessa regione nella madre sana. (C) Pedigree della famiglia: il padre ed entrambi i figli, in nero, hanno una mutazione confermata in GDF5. La madre sana appare in bianco. Il quadrato grigio rappresenta uno zio paterno con polidattilia bilaterale post-assiale che probabilmente ha la mutazione.
Il fattore 5 di differenziazione della crescita (GDF5) è strettamente associato alle proteine morfogenetiche ossee e appartiene alla superfamiglia del fattore di crescita trasformante β, ed è coinvolto nello sviluppo scheletrico e articolare embrionale.4 Il gene GDF5 è un hotspot mutazionale per disturbi associati a malformazioni scheletriche.5 La maggior parte delle mutazioni omozigoti o eterozigoti composti sono associati a malattie gravi: Condrodisplasia di tipo Grebe (OMIM 200700), displasia acromesomelica di tipo Hunter-Thomson (OMIM 201250) o sindrome di Du Pan (OMIM 228900). D’altra parte, mutazioni eterozigote associate a displasie scheletriche più lievi: simpalangismo prossimale 1 B (OMIM 615298) e sindrome da sinostosi multipla tipo 2 (OMIM 610017), entrambe associate a mutazioni missenso con guadagno di funzione, e brachidattilia tipo A1 e A2, anch’esse associate a mutazioni missenso, ma con perdita di funzione.5 La brachidattilia di tipo C è associata a mutazioni eterozigoti con perdita di funzione, sebbene siano stati riportati anche 3 casi con eredità recessiva.6 La maggior parte delle mutazioni associate alla BDC sono mutazioni frameshift nella parte del prodominio del gene, mentre la maggior parte delle mutazioni nel dominio maturo sono mutazioni missenso, con un’espressione fenotipica altamente variabile.4
La famiglia che presentiamo qui ha una mutazione non senso nella regione che codifica per il dominio attivo della proteina, con conseguente eliminazione dei suoi ultimi 14 aminoacidi. Questa è la seconda mutazione senza senso che colpisce il dominio attivo maturo descritto in letteratura.3 La prima è una mutazione simile nell’aminoacido immediatamente precedente a quello mutato nella famiglia che descriviamo qui (p.Tyr487*/c.1461T>G), il che suggerisce che entrambi danno origine a monomeri mutanti e aploinsufficienza funzionale di GDF5, causando così la BDC.
Finanziamento
Questo studio è stato parzialmente finanziato dai progetti PI13/00467 e PI13/01295, integrati nel piano R&D&I 2013-2016 del governo spagnolo e cofinanziati dal Vice Direttore Generale di Valutazione e Promozione della Ricerca dell’Instituto de Salud Carlos III (ISCIII), il Fondo Europeo di Sviluppo Regionale (FESR) e il Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición (Centro in rete di ricerca biomedica sulla fisiopatologia dell’obesità e della nutrizione), ISCIII, Madrid.