Tecknet brakydaktyli omfattar en grupp av bendysplasier som involverar handens och fötternas falanger och/eller metakarpala/metatarsala ben. Det finns 5 typer (A-E) och flera subtyper (A1-A4; E1-E3). Den har ett autosomalt dominant ärftlighetsmönster med varierande penetrans. Fall med recessivt arv är extremt sällsynta.
Brachydaktyli typ C (BDC) kännetecknas av en förkortning av de mellersta falangerna i andra, tredje och femte fingret och det första metakarpalbenet, och kan också uppvisa en ulnaravvikelse av pekfingret, polydaktyli eller en distinkt hypersegmentering av de proximala eller mellersta falangerna i andra och tredje fingret. Fjärde fingret är minst påverkat och vanligtvis längst.1-3 ”Ängelformade” falanger (Fig. 1A) är visserligen karakteristiska för BDC, men är inte patognomoniska, eftersom de också kan förekomma i den sjukdom som kallas ängselformad phalango-epifyseal dysplasi. Det är möjligt att denna sjukdom och BDC är en del av samma kliniska spektrum.1 I BDC normaliseras detta särdrag när den fysiska förslutningen av handbenen är avslutad och slutar som enkel brakydaktyli.1 Andra anomalier som förknippas med BDC är kortväxthet med fördröjd benålder, Madelungdeformitet, höftledsdysplasi, talipes valgus eller equinovarus eller avsaknad av mellanfingrar i tårna.2,3
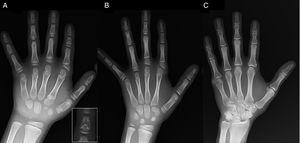
(A) Röntgenbild av den vänstra handen hos proband (kronologisk ålder, 7 år; benålder, 5 år), med förkortning av ett onormalt första metacarpal (dubbla proximala och distala epifysplattor) och de mellersta falangerna i det andra, tredje och femte fingret. Andra fingret uppvisar ulnaravvikelse, och fjärde fingret är minst påverkat och längst i vänster hand. De proximala epifyserna i andra och tredje fingret är dysplastiska, med en iögonfallande ängelformad mittfals i andra fingret (infälld i A). (B). Röntgenbild av den vänstra handen hos probandens syster (5½ år gammal utan fördröjning av benåldern), med förkortning av andra, tredje och femte fingrets mittfingertoppar och ett normalt fjärde finger. I detta fall var den första mellanhanden normal. Det mest framträdande kännetecknet är den triangulära formen på den proximala epifysen på andra fingrets proximala falang, som liknar broderns, och den trapetsformiga formen på falangen i mittfingret. Liksom hennes bror uppvisar hennes andra finger ulnardeviation. (C) Röntgenbild av den vänstra handen hos fadern till den undersökta personen, som endast visar en benig rest av en postaxial hexadaktyli som korrigerades kirurgiskt i barndomen.
Vi presenterar fallet med en pojke i åldern 7 år med radiologiska kännetecken som var förenliga med BDC och som remitterades för utvärdering av kortväxthet (z-score, -1,8). Hans far hade unilateral postaxial polydaktyli, som också fanns bilateralt hos en farbror till fadern. Radiografisk undersökning av vänster hand och handled avslöjade en benålder som var 2 år yngre än den kronologiska åldern och anomalier som föranledde en skelettundersökning. Undersökningen påvisade anomalier i händerna (fig. 1A) och subtila avvikelser i fötterna (mild epifysedysplasi i de proximala falangerna på några tår). Den radiologiska utvärderingen av en syster som var 6 år gammal avslöjade skador i handen som liknade dem som fanns hos den provade, även om de var mindre uttalade (fig. 1B). En röntgenbild av faderns hand (Fig. 1C) visade endast en benrest av den postaxiala hexadaktyli som hade korrigerats kirurgiskt i barndomen.
Sekvensering av GDF5-genen hos probandens visade en heterozygot punktsubstitution i exon 2 (c.1462A>T) som resulterar i en för tidig stoppkodon (p.Lys488*, nonsensmutation) och ett trunkerat protein som är 14 aminosyror kortare än det vilda proteinet (Fig. 2A). Denna mutation hittades också hos patientens far och syster, men inte hos den friska modern (Fig. 2B). Fig. 2C visar familjens stamtavla. Detta är en ny variant som troligen är patogen och med ett autosomalt dominant arvsmönster.
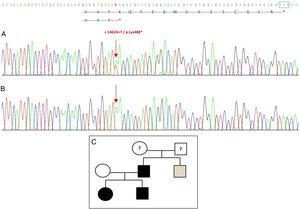
Novell mutation som upptäckts i GDF5-genen. (A) Gensekvens för exon 2 hos proband, som visar en c.1462A>T-mutation som resulterar i ett för tidigt stoppkodon och ett trunkerat protein (p.Lys488*) (vi har angett stoppkodonets normala position med en grön kvadrat). (B) Normal sekvens av samma region hos den friska modern. (C) Familjens stamtavla: fadern och båda barnen, i svart, har en bekräftad mutation i GDF5. Den friska modern visas i vitt. Den grå kvadraten representerar en farbror till fadern med bilateral postaxial polydaktyli som troligen har mutationen.
Växtdifferentieringsfaktor 5 (GDF5) är nära förknippad med benmorfogenetiska proteiner och tillhör den transformerande tillväxtfaktorn β superfamiljen, med är involverad i embryonal skelett- och ledutveckling.4 GDF5-genen är en mutationell hotspot för sjukdomar som är förknippade med skelettmissbildningar.5 De flesta homozygota eller sammansatta heterozygota mutationer är förknippade med allvarliga sjukdomar: De flesta av dessa mutationer är dödliga och heterotermiska, men de leder till allvarliga sjukdomar, t.ex. chondrodysplasi av Grebe-typ (OMIM 200700), akromesomelisk dysplasi av Hunter-Thomson-typ (OMIM 201250) eller Du Pan-syndromet (OMIM 228900). Å andra sidan är heterozygota mutationer förknippade med mildare skelettdysplasier: proximal symphalangism 1 B (OMIM 615298) och multipelt synostosis syndrom typ 2 (OMIM 610017), som båda är förknippade med missense-mutationer med funktionsförstärkning, och brakydaktyli typ A1 och A2, som också är förknippade med missense-mutationer men med funktionsförlust.5 Brakydaktyli typ C är associerad med heterozygota mutationer med funktionsförlust, även om tre fall med recessiv nedärvning också har rapporterats.6 De flesta mutationer som förknippas med BDC är frameshift-mutationer i prodomändelen av genen, medan de flesta mutationer i den mogna domänen är missense-mutationer, med ett mycket varierande fenotypiskt uttryck.4
Familjen som vi presenterar här har en nonsens-mutation i den region som kodar för proteinets aktiva domän, vilket resulterar i att dess sista 14 aminosyror elimineras. Detta är den andra nonsensmutationen som påverkar den aktiva mogna domänen och som beskrivs i litteraturen.3 Den första är en liknande mutation i aminosyran omedelbart före den som är muterad i den familj som vi beskriver här (p.Tyr487*/c.1461T>G), vilket tyder på att båda ger upphov till muterade monomerer och funktionell haploinsufficiens av GDF5 och därmed orsakar BDC.
Finansiering
Denna studie finansierades delvis av projekten PI13/00467 och PI13/01295, integrerade i den spanska regeringens R&D&I-plan 2013-2016 och samfinansierade av Deputy General-Directorate of Research Evaluation and Promotion of the Instituto de Salud Carlos III (ISCIII), Europeiska regionala utvecklingsfonden (ERUF) och Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición (Centrum för nätverk för biomedicinsk forskning om fysiopatologi vid fetma och nutrition), ISCIII, Madrid.